- Progeria
-
Progeria 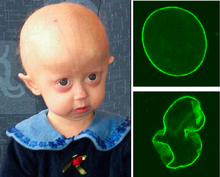
Niño con Sindrome de ProgeriaClasificación y recursos externos CIE-10 E34.8 CIE-9 259.8 PubMed Buscar en Medline mediante PubMed (en inglés) eMedicine derm/731 MeSH D011371 Sinónimos Síndrome de Werner. Envejecimiento prematuro. Envejecimiento precoz. Progeria del adulto.  Aviso médico
Aviso médico Progeria (del griego geras, "vejez") es una enfermedad genética de la infancia extremadamente rara, caracterizada por un envejecimiento brusco y prematuro. Se estima que afecta a uno de cada 8 millones de recién nacidos. La progeria puede afectar diferentes órganos y tejidos: hueso, músculos, piel, tejido subcutáneo y vasos.
La forma más severa de esta enfermedad es la llamada síndrome de Hutchinson-Gilford nombrada así en honor de Jonathan Hutchinson, quién fue el primero en describirla en 1886 y de Hastings-Gilford quien realizó diferentes estudios acerca de su desarrollo y características en 1904.
Contenido
Características clínicas
- Baja estatura
- Piel seca y arrugada
- Calvicie prematura
- Canas en la infancia
- Ojos prominentes
- Cráneo de gran tamaño
- Venas craneales sobresalientes
- Ausencia de cejas y pestañas
- Nariz grande y con forma de pico
- Mentón retraído
- Problemas cardíacos
- Pecho angosto, con costillas marcadas
- Extremidades finas y esqueléticas
- Estrechamiento de las arterias coronarias
- Articulaciones grandes y rígidas
- Manchas en la piel semejantes a las de la vejez por mal metabolismo de la melanina
- Presencia de enfermedades degenerativas como la artritis o cataratas, propias de la vejez
- Muerte natural hacia los 13 años.
- Mitosis con retardo reticoendoplasmatico.
- Falta de gametos sexuales y pensativos.
Causa
La progeria está reconocida como una laminopatía, asociada a mutaciones en el gen LMNA que codifica para la lamina A/C, el componente principal de las laminas nucleares. La mutación más frecuente es una mutación puntual en la posición 1824 en el exón 11, que crea una mutación en el codón 608 y activa el sitio críptico de splice llevando a una lamina A truncada. Como consecuencia, se produce la pérdida de 50 aminoácidos en el terminal-C de la forma de la proteína conocida como progerina o lamina AD50. Esto lleva a la disrupción del ensamblaje normal de la envoltura nuclear, la función nuclear y la función de la lamina A. Afecta específicamente la maduración de la prelaminina A a la laminina A; por lo tanto, la progeria es un desorden que tiene un efecto profundo en la integridad del tejido conectivo. Esto es crítico para el soporte nuclear y para la organización de la cromatina. Teniendo en cuenta lo anterior, los estudios se han basado en fibroblastos, ya que la enfermedad se manifiesta en el tejido conectivo. Se han encontrado cambios en la glicosilación de los fibroblastos, pero aún no se sabe si esto se debe a algún estado de la enfermedad, o a la adquisición de mutaciones genómicas.
Las células presentan un núcleo con alteraciones estructurales (herniaciones y lóbulos) así como defectos en la organización de la heterocromatina. Molecularmente presentan un defecto en el mecanismo de reparación del ADN como consecuencia de la rotura de la hélice doble.
Diagnóstico
Al nacer, los niños con progeria parecen normales aunque esclerodermas, cianosis facial y nariz esculpida pueden ser aparentes. Los síntomas se manifiestan durante el primer año, con uno o con varios de las siguientes anomalías: retardo en el crecimiento, alopecia, anomalías en la piel.
Pronóstico
El promedio de vida en niños enfermos es de 13 años, pero puede estar entre 7-45 años, aunque la supervivencia más allá de la adolescencia es inusual. En más del 80% de los casos la muerte se debe a complicaciones que surgen, como la ateroesclerosis, fallos en el corazón, infarto del miocardio y trombosis coronaria.
Tratamiento
No existe aún un tratamiento de probada eficacia. La mayoría de los tratamientos se limitan a paliativos o prevención de complicaciones como son las enfermedades cardiovasculares. Se utilizan aspirinas en bajas dosis y dietas hipercalóricas. Se han intentado tratamientos con hormona de crecimiento humano.
Después de ser descubierto el gen causante de ella y su mecanismo se ha propuesto un tratamiento con un tipo de droga anticancerígena, inhibidora de la farnesyltransferasa (FTIs), se ha probado su eficacia en modelos con ratones.
A partir de Mayo de 2007 se inició un período de pruebas clínicas con pacientes utilizando FTI Lonafarnib.
Aunque recientemente se ha descubierto específicamente el gen causante de la progeria, aún no existe cura.
Véase también
Referencias
O´Brien, Martin; Jensen, Sacha; Weiss, Anthony (1998). «Hutchinson-Gilford progeria: faithful DNA maintenance, inheritance and allelic trancription of b(1-4) galactosyltransferase». Mechanisms of Aging and Development 101. 43-56.
Pilarczyka, Agnieszka; Kmiec, Tomasz; et al (2008). «Progeria caused by rare LMNA mutation p.S143F associated with mild myopathy and atrial fibrillation». European Journal of Pediatric Neurology 12. 427-430.
Lemire, Joan; Patis, Carris; et al (2006). «Aggrecan expression is substantially and abnormally upregulated in Hutchinson-Gilford Progeria Syndrome dermal fibroblasts». Mechanisms of Aging and Development 127. 660-669.
Enlaces externos
Categorías:- Enfermedades genéticas
- Enfermedades raras
- Gerontología
Wikimedia foundation. 2010.