- Enfermedad de von Willebrand
-
Enfermedad de von Willebrand
Enfermedad de von Willebrand
Clasificación y recursos externos Aviso médico
Aviso médicoCIE-10 D 68 0; d 65 CIE-9 286.4 OMIM 193400 DiseasesDB 14007 Medline Buscar en Medline (en inglés) MeSH D014842 Sinónimos La enfermedad de von Willebrand (EvW) es la anomalía en la coagulación de carácter hereditaria más común entre los humanos, aunque también puede ser adquirida como consecuencia de otras condiciones médicas. Se debe a una deficiencia cualitativa o cuantitativa del factor de von Willebrand (FvW), una proteína multimérica requerida para la agregación plaquetaria. Hay cuatro tipos de EvW y se sabe que afecta a seres humanos y perros. Otros factores, como los grupos sanguíneos ABO, también pueden desempeñar un papel en el grado de la enfermedad. Su nombre se debe a Erik Adolf von Willebrand (1870–1949), un pediatra finlandés que descubrió la enfermedad en 1926.[1]
Contenido
Diagnóstico
Los diversos tipos de EvW se presentan con distintos grados de hemorragia, generalmente en forma de dolor, sangrado nasal y sangrado de encías. Las mujeres pueden experimentar períodos menstruales pesados y pérdida de sangre durante el parto. Las hemorragias internas o conjuntas de carácter grave son poco frecuentes, ocurriendo sólo en el tipo 3 de EvW.
Cuando se sospecha, el plasma sanguíneo de un paciente debe ser investigado por las posibles deficiencias cuantitativas y cualitativas de FvW. Esto se logra midiendo la cantidad de FvW en un ensayo de antígeno FvW y la funcionalidad de FvW con un ensayo de glicoproteína vinculante (GPIb), un ensayo de colágeno vinculante o ensayos de actividad del cofactor ristocetin o de aglutinación plaquetaria inducida por ristocetin. Los niveles de factor VIII de coagulación también se estudian porque éste se une al FvW, que protege el factor VIII de la rápida distribución dentro de la sangre. La deficiencia de FvW, por tanto, puede dar lugar a una reducción en los niveles de factor VIII. Los niveles normales no excluyen todas las formas de EvW: particularmente el tipo 2, que sólo puede ser revelado investigando la interacción de las plaquetas con subendothelium bajo flujo (PAF), un estudio altamente especializado de coagulación no realizado de forma rutinaria en la mayoría de laboratorios clínicos. Un ensayo de agregación plaquetaria mostrará una respuesta anormal a ristocetin con respuestas normales a otros agonistas utilizados. Un ensayo de función plaquetaria dará un tiempo de cierre anormal de colágeno y adrenalina y en la mayoría de los casos (pero no en todos) un tiempo normal de colágeno. El tipo 2N sólo puede ser diagnosticado mediante la realización de un ensayo de factor VIII vinculante. La detección de la enfermedad es complicada debido a que el FvW es un reactivo de fase aguda con el aumento de niveles en infección, embarazo y estrés.
Otras pruebas realizadas a cualquier paciente con problemas de sangrado son un recuento sanguíneo completo (especialmente de plaquetas), TTPA, tiempo de protrombina, tiempo de trombina y nivel de fibrina. También pueden llevarse a cabo las pruebas para el factor IX si se sospecha de hemofilia B. Los pacientes con EvW mostrarán, por lo general, un tiempo de protrombina normal y una prolongación variable del tiempo de tromboplastina parcial.
- Adquisición de la enfermedad
La adquisición de la enfermedad puede ocurrir en pacientes con autoanticuerpos. En este caso, la función del FvW no es inhibida pero el complejo anticuerpo-FvW es eliminado rápidamente de la circulación. Una forma de EvW se produce en pacientes con estenosis aórtica, lo que lleva a una hemorragia gastrointestinal (síndrome de Heyde). Esta forma de adquisición puede ser más frecuente de lo comúnmente pensado. La adquisición de EvW también se ha descrito en los siguientes trastornos: tumor de Wilms, hipotiroidismo y displasia mesenquimal.
Clasificación y tipos
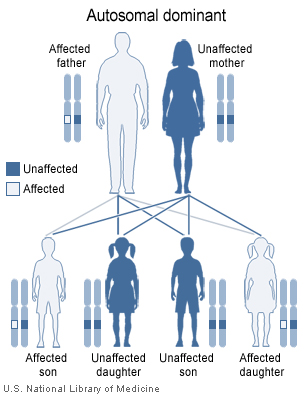
 Los tipos 1 y 2 son heredados como dominante.
Los tipos 1 y 2 son heredados como dominante.
 El tipo 3 (y en ocasiones el 2) se hereda como recesivo.
El tipo 3 (y en ocasiones el 2) se hereda como recesivo.Hay cuatro tipos hereditarios de la enfermedad: tipo 1, tipo 2, tipo 3 y tipo plaquetas. La mayoría de los casos son hereditarios, pero se han dado formas adquiridas de la enfermedad. La clasificación de la Sociedad Internacional de Trombosis y Hemostasis depende de la definición de los defectos cualitativos y cuantitativos.[2]
Tipo 1
El tipo 1 de EvW (60-80% de los casos) es un defecto cuantitativo (heterocigotos para el gen defectuoso) pero puede no tener perjudicada la coagulación, llevando la mayoría de los pacientes una vida casi normal. Los problemas pueden surgir en forma de sangrado después de una intervención quirúrgica (incluidos los procedimientos dentales), dolor intenso o menorragia. La disminución de los niveles de FvW son detectados (10-45% de lo normal).
Tipo 2
El tipo 2 de EvW (20-30%) es un defecto cualitativo y la tendencia de sangrado puede variar entre individuos. Hay niveles normales de FvW pero los oligómeros son estructuralmente anormales o subgrupos de grandes o pequeños oligómeros están ausentes. Existen cuatro subtipos:
- Tipo 2A
Se trata de una alteración de la síntesis o proteólisis de oligómeros de FvW dando resultado a la presencia de pequeñas unidades en circulación. El factor VIII vinculante es normal. Tiene una baja y desproporcionada actividad del co-factor ristocetin en comparación con el antígeno de von Willebrand.
- Tipo 2B
Este es un defecto de "ganancia de función", llevando a una espontánea unión a las plaquetas y la posterior limpieza de las plaquetas y los oligómeros del FvW. Puede ocurrir una leve trombocitopenia. Los oligómeros del FvW están ausentes en la circulación y el factor VIII vinculante es normal. Al igual que en el tipo 2A, el co-factor ristocetin es bajo cuando el plasma pobre de plaquetas del paciente es ensayado con donantes de plaquetas normales. Sin embargo, cuando el ensayo se realiza con las plaquetas del propio paciente (plasma rico en plaquetas), una cantidad de ristocetin menor de la normal causa que se produzca agregación. Esto es debido a los oligómeros del FvW restantes ligados a las plaquetas del paciente. Los pacientes con este subtipo no pueden utilizar la desmopresina como tratamiento contra el sangrado ya que puede dar lugar a una agregación plaquetaria no deseada.
- Tipo 2M
Este tipo es causado por la disminución o ausencia de unión a GPIb sobre las plaquetas. El factor VIII vinculante es normal.
- Tipo 2N
Esta es una deficiencia de la unión del FvW y el factor VIII. Este tipo da un nivel normal de antígeno FvW y resultados de la prueba normales, pero tiene un bajo factor VIII. Esto ha llevado a que algunos pacientes de 2N fueran diagnosticados en el pasado como hemofilia A, debiendo ser sospechosos si el paciente tiene los hallazgos clínicos de la hemofilia A pero sugiere una filiación autosómica no ligada al cromosoma X, la herencia.
Tipo 3
El tipo 3 es la forma más grave de la enfermedad (homocigótica para el gen defectuoso) y puede tener graves hemorragias de mucosas, antígeno de FvW no detectable y suficientemente bajo el factor VIII que puede ocasionar hemartrosis (sangrado conjunto), como en casos de leve hemofilia.
Tipo plaquetas
El tipo plaquetas (también conocido como pseudo-EvW) es dominante autosómico causado por mutaciones de ganancia de función del receptor de FvW en plaquetas; específicamente, la cadena alfa de la glicoproteína del receptor. Esta proteína es parte del complejo que forma el pleno receptor de FvW en las plaquetas. La actividad de ristocetin y la pérdida de oligómeros de FvW son similares al tipo 2B, pero las pruebas genéticas de FvW no revelan mutaciones.
Fisiología
El FvW es principalmente activo en condiciones de alto flujo sanguíneo y tensión cortante. Por tanto, la deficiencia del factor se muestra sobre todo en órganos con pequeños vasos como la piel, el aparato digestivo y el útero. En la angiodisplasia, una forma de telangiectasia del colon, la tensión cortante es mucho mayor que en el promedio de los capilares y el riesgo de sangrado aumenta.
En los casos más graves del tipo 1 son comunes los cambios genéticos en el gen del FvW y son altamente penetrantes. En los casos más leves de este tipo, puede haber un complejo espectro de patología molecular, además de los polimorfismos del gen de FvW solo.[3] El sistema ABO del individuo puede influir en la presentación y patología de la enfermedad. Aquellos individuos con grupo sanguíneo O tienen un nivel medio menor al de personas con otros grupos sanguíneos. A menos que el antígeno de FvW específico del grupo ABO referencie rangos habituales, los individuos del grupo O pueden ser diagnosticados como de tipo 1 y algunas personas de grupo sanguíneo AB con un defecto genético de FvW pueden pasar por alto el diagnóstico porque los niveles son elevados debido al grupo sanguíneo.[4]
Genética
El gen del FvW se localiza en el cromosoma doce (12p13.2). Tiene 52 exónes que abarcan 178kbp. Los tipos 1 y 2 se heredan como dominantes y el tipo 3 se hereda como recesivo. Ocasionalmente, el tipo 2 también se hereda de forma recesiva.
Epidemiología
La prevalencia de la enfermedad es de aproximadamente una de cada cien personas.[5] Sin embargo, la mayoría de esas personas no presentan síntomas. La prevalencia de casos significativos clínicamente es de cien por millón.[5] Debido a que buena parte de las formas son más bien leves, se detectan con mayor frecuencia en mujeres, cuya tendencia al sangrado se muestra durante la menstruación. Puede ser más severa o aparente en personas con sangre tipo O.
Terapia
Normalmente, los pacientes con EvW no requieren un tratamiento regular, aunque siempre están en mayor riesgo por hemorragia. Para las mujeres con un sagrado menstrual abundante, la combinación de píldoras anticonceptivas orales puede ser eficaz en la reducción del sangrado o en la reducción de la duración o frecuencia de los períodos. A veces se da un tratamiento profiláctico a pacientes con EvW que tienen programada una intervención quirúrgica; pueden ser tratados con factor VIII concentrado completando al FvW (factor antihemofílico, más comúnmente conocido como Humate-P), y otros casos leves pueden ser probados con desmopresina (1-desamino-8-D-arginina vasopresina), que trabaja para elevar los niveles de plasma del FvW del propio paciente induciendo la liberación de FvW almacenado en el cuerpo de Weibel-Palade en las células endoteliales.
Véase también
- Factor de von Willebrand
- Hematología
- Hemofilia
- Portal:Medicina
Referencias
- ↑ Who named it?. «Erik Adolf von Willebrand» (en inglés). Consultado el 4 de agosto de 2009.
- ↑ (1994) Thromb. Haemost., volumen 71, tema 4, páginas 520-5 (ed.). A revised classification of von Willebrand disease. For the Subcommittee on von Willebrand Factor of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. PMID 8052974.
- ↑ James P., Notley C., Hegadorn C., Leggo J., Tuttle A., Tinlin S., Brown C., Andrews C., Labelle A., Chirinian Y., O'Brien L., Othman M., Rivard G., Rapson D., Hough C., Lillicrap D. (2007). Blood, volumen 109, tema 1, páginas 145-54 (ed.). The mutational spectrum of type 1 von Willebrand disease: Results from a Canadian cohort study. PMID 17190853.
- ↑ Gill JC., Endres-Brooks J., Bauer PJ., Marks WJ., Montgomery RR. (1987). Blood, volumen 69, tema 6, páginas 1691-5 (ed.). The effect of ABO blood group on the diagnosis of von Willebrand disease. PMID 3495304 .
- ↑ a b (septiembre de 2004) Haematologica, volumen 89, tema 9, página 1036 (ed.). Molecular basis of von Willebrand disease and its clinical implications. PMID 15377463.
Bibliografía
-
- Sadler, J.E. (1998). Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem., volumen 67, páginas 395-424. PMID 9759493.
- Mannucci P.M. (2004). Treatment of von Willebrand's Disease. N. Engl. J. Med., volumen 351, tema 7, páginas 683-94. PMID 15306670.
- Laffan M., Brown S.A., Collins P.W., y otros (2004). The diagnosis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors' Organization. Haemophilia, volumen 10, tema 3, páginas 199-217. PMID 15086318.
Enlaces externos
- Enfermedad de von Willebrand
- Guía clínica de la enfermedad (en inglés)
- Federación Mundial de Hemofilia
- The Haemophilia Society (en inglés)
- National Hemophilia Foundation (en inglés)
- Base de datos sobre el FvW (en inglés)
Categorías: Enfermedades epónimas | Enfermedades hematológicas | Enfermedades hereditarias

Wikimedia foundation. 2010.