- Adrenoleucodistrofia
-
Adrenoleucodistrofia Clasificación y recursos externos CIE-10 E71.3 CIE-9 330.0 OMIM 300100 DiseasesDB 292 MeSH D000326  Aviso médico
Aviso médico La adrenoleucodistrofia (ALD) es una enfermedad hereditaria incluida en el grupo de las leucodistrofias. Produce una desmielinización intensa y la muerte prematura en niños, y la adrenomieloneuropatía se asocia a una neuropatía mixta, motora y sensorial, con paraplejía espástica en los adultos. Ambos procesos cursan con niveles circulantes elevados de ácidos grasos de cadenas muy largas que provocan insuficiencia suprarrenal.
Esta enfermedad se caracteriza por la presencia de una degeneración progresiva de la corteza suprarrenal y motora, lo que da lugar a una insuficiencia suprarrenal o Enfermedad de Addison, asociada a la desmielinización de la sustancia blanca del sistema nervioso central (sistema formado por el encéfalo y la médula espinal), con pérdida de la cubierta de mielina (vaina de sustancia blanca que recubre los nervios) de un tipo de fibras nerviosas del cerebro.
Contenido
Tipos
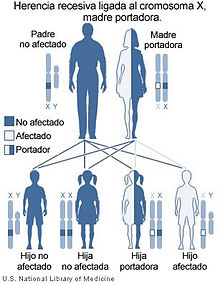 Modelo de herencia de un carácter recesivo ligado al sexo, como la ALD.
Modelo de herencia de un carácter recesivo ligado al sexo, como la ALD.
Existen diferentes tipos de adrenoleucodistrofia, en función de la edad de comienzo:
- Adrenoleucodistrofia neonatal. Forma heredada como un rasgo genético recesivo, que comienza típicamente durante los primeros meses de la vida o período neonatal. Los lactantes comienzan con deterioro neurológico y presentan o desarrollan signos de disfunción de la corteza suprarrenal. Casi todos los pacientes sufren retraso mental y fallecen antes de los 5 años de edad.
Tratamiento
Un tratamiento con diferentes aceites ("aceite de Lorenzo") ha tenido mucho éxito aunque no ha sido aprobada por la FDA. Se trata de una mezcla de ácidos grasos que reduce los niveles de ácidos grasos de cadena muy larga, los cuales son la causa principal de la ALD, por medio de la competitividad, inhibiendo la enzima que forman los ácidos grasos de cadena muy larga.
El New England Journal of Medicine reportó que en unas pruebas con varios individuos que sufrían de adrenomieloneuropatía no encontraron evidencias satisfactorias. Sin embargo, en el trabajo de Moser y cols. (entre ellos el propio Odone)[1] se recoge que de 89 niños diagnosticados de ALD-X que eran asintomáticos y que empezaron a tomar el aceite de Lorenzo en ese instante, tan sólo en un 24% de los casos terminaron desarrollando anomalías recogidas en resonancia magnética y en un 11% anomalías de neuroimagen y neurológicas, con control del deterioro en el resto de los niños (76 y 89%, respectivamente). Tan alto porcentaje de control de los signos clínicos de la enfermedad, ineludiblemente llevan a la conclusión de la efectividad del aceite de Lorenzo; efectividad que nunca se consigue al 100% con ninguna clase de fármaco, medicamento o tratamiento.
Referencias
- ↑ Moser, HW; Raymond GV, Lu S-E, Muenz LR, Moser AB, Xu J, Jones RO, Loes DJ, Melhem ER, Dubey P, Bezman L, Brereton NH, Odone A (2005-07). «Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzo's Oil.». Archives of Neurology 62 (7): pp. p. 1073–80. doi:. PMID 16009761.
Enlaces externos
Categorías:- Enfermedades hereditarias
- Enfermedades metabólicas
- Leucodistrofias
- Glándula suprarrenal
Wikimedia foundation. 2010.