- Tirosinemia tipo 1
-
Tirosinemia tipo 1 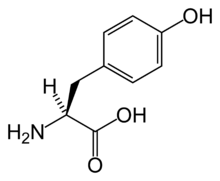
Clasificación y recursos externos CIE-10 E70.2 CIE-9 270.2 OMIM 276700 DiseasesDB 13478 eMedicine ped/2339 MeSH D020176  Aviso médico
Aviso médico La tirosinemia tipo I, también conocida como tirosinemia hepatorenal, es la forma más severa de tirosinemia. Es causada por la deficiencia de la enzima fumarilacetoacetato hidrolasa.
Contenido
Genética
La tirosinemia tipo I tiene herencia autosómica recesiva. A nivel mundial,la tirosinemia tipo 1 afecta alrededo de 1 persona en 100.000 habitantes. Este tipo de tirosinemia es mucho más común en Quebec, Canadá. La incidencia total en este lugar es alrededor de 1 en 16.000 habitantes. En la región de Saguenay–Lac-Saint-Jean, de Quebec, la tirosinemia tipo 1 afecta a 1 persona en 1846 habitantes[1] The carrier rate has been estimated to be between 1 in 20 and 1 in 31.[2]
Patofisiología
La fumarilacetoacetato hidrolasa cataliza la etapa final de la degradación de la tirosina- fumarilacetoacetato a fumarato, acetoacetato y succinato. El fumarilacetoacetato se acumula en los hepatocitos y en las células del túbulo proximal del riñón, causando estrés oxidativo y daño a nivel del ADN, conduciendo a la muerte celular y la disfunción en la expresión genética la cual altera procesos metabólicos como la síntesis protéica y la gluconeogénesis. El incremento del fumarilacetoacetato inhibe los pasos previos de la degradación de la tirosina, lo cual también conduce a la acumulación de tirosina en el cuerpo. La tirosina no es directamente tóxica para el hígado ni para los riñones pero causa problemas a nivel dermatológico y neurológico.
Signos y síntomas
Este tipo de tirosinemia típicamente presenta falla hepática acompañado de hepatomegalia. Los efectos primarios son progresivos, provocando también disfunción renal. La enfermedad hepática produce cirrosis, hiperbilirrubinemia directa, aumento de alfa feto proteína, hipoglicemia y alteraciones de la coagulación. Puede producir [ictericia]], asciti y hemorragia. Existe además un aumento en el riesgo de hepatoarcinoma. La falla renal presenta síndrome de Fanconi: acidosis tubular renal, hipofosfatemia y aminoaciduria. La cardiomiopatía, y alteraciones neurológicas y dermatológicas también pueden ser observadas.
Tratamiento
El tratamiento primario para la tirosinemia tipo 1 es Nitisinona (orfadin). Nitisinona inhibe la conversión de 4-OH fenilpiruvato a ácido homogenistisico por la 4-OH fenilpiruvato dioxigenasa, el segundo paso de la degradación de la tirosina. Al inhibir esta enzima,se previene la acumulación de fumarolacetoacetato. Anteriormente, el trasplante de hígado era el tratamiento de elección y aún sigue siendo usado en aquellos pacientes en donde falla el manejo farmacológico.
Referencias
- ↑ Grompe M, St-Louis M, Demers SI, al-Dhalimy M, Leclerc B, Tanguay RM (1994). «A single mutation of the fumarylacetoacetate hydrolase gene in French Canadians with hereditary tyrosinemia type I». N. Engl. J. Med. 331 (6): pp. 353–7. doi:. PMID 8028615. http://content.nejm.org/cgi/pmidlookup?view=short&pmid=8028615&promo=ONFLNS19.
- ↑ Laberge, C.; L. Dallaire (October 28, 1967). «Genetic aspects of tyrosinemia in the Chicoutimi region». Can Med Assoc J 97 (18): pp. 1099–1101. PMID 6057677.
Categorías:- Enfermedades metabólicas
- Errores congénitos del metabolismo
Wikimedia foundation. 2010.