- Enfermedad de Batten
-
Enfermedad de Batten 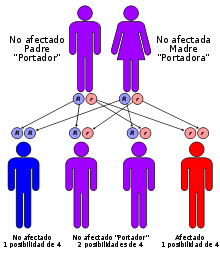
Herencia mendeliana autosómica recesiva: dos mutaciones de línea germinal (una de cada uno de los padres) para desarrollar la enfermedad; igualmente transmitida por hombres y mujeres.Clasificación y recursos externos CIE-10 E75.4 CIE-9 330.1 OMIM 204200 DiseasesDB 31534 PubMed Buscar en Medline mediante PubMed (en inglés) Sinónimos Enfermedad de Spielmeyer-Vogt-Sjogren-Batten.  Aviso médico
Aviso médico La enfermedad de Batten es una enfermedad hereditaria mortal que afecta al sistema nervioso y que comienza en la niñez. Los primeros síntomas de este trastorno aparecen generalmente entre las edades de 5 y 10 años, cuando los padres o los médicos advierten que un niño previamente normal ha comenzado a presentar convulsiones o problemas de visión. En algunos casos los primeros signos son sutiles, manifestándose en cambios de personalidad y del comportamiento, lentitud en el aprendizaje, torpeza o tropiezos al caminar.
Al pasar del tiempo, los niños afectados padecen incapacidades mentales, convulsiones más severas y la pérdida progresiva de la vista y de las capacidades motrices. Los niños que padecen la enfermedad de Batten terminan quedando ciegos, postrados en una cama y hasta dementes. La enfermedad de Batten a menudo es mortal al llegar a los últimos años de la adolescencia o al llegar a la edad de 20 años.
La enfermedad de Batten recibe su nombre del pediatra británico que la describió por primera vez en 1903. También conocida como enfermedad de Spielmeyer-Vogt-Sjogren-Batten, es la forma más común de un grupo de trastornos llamados ceroidolipofuscinosis neuronales (NCL por su sigla en inglés). Aunque la enfermedad de Batten se considera generalmente como la forma juvenil de NCL, algunos médicos suelen utilizar el término de enfermedad de Batten para describir todas las formas de ceroidolipofuscinosis. Se incluye dentro de las lipidosis o enfermedades por almacenamiento de lípidos.
Contenido
Otras formas de ceroidolipofuscinosis neuronal
Existen otros tres tipos principales de ceroidolipofuscinosis neuronales (NCL), incluyendo dos formas que empiezan al principio de la niñez y una forma muy poco común que afecta a los adultos. Los síntomas de estos tres tipos son similares a los causados por la enfermedad de Batten, pero llegan a ser evidentes en diversas edades y progresan en modos distintos.
- La ceroidolipofuscinosis neuronal infantil (enfermedad de Santavuori-Haltia) comienza cerca de los 6 meses a los 2 años de edad y progresa rápidamente. Los niños afectados no suelen desarrollarse y presentan una cabeza anormalmente pequeña (microcefalia). También son típicas las contracciones cortas y agudas de los músculos llamadas espasmos mioclónicos. Los pacientes mueren generalmente antes de la edad 5 años, aunque algunos han permanecido en estado vegetativo algunos años más.
- La ceroidolipofuscinosis neuronal infantil tardía (enfermedad de Jansky-Bielschowsky) comienza entre las edades de 2 y 4 años. Los signos precoces típicos son la pérdida de coordinación de los músculos (ataxia) y convulsiones que no responden a los medicamentos. Esta forma progresa rápidamente y termina siendo letal en las edades entre 8 y 12 años.
- La ceroidolipofuscinosis neuronal del adulto (enfermedad de Kufs o la enfermedad de Parry) comienza generalmente antes de la edad de 40 años, causa síntomas más leves que progresan lentamente y no causa ceguera. Aunque la edad de la muerte es variable entre los pacientes afectados, este trastorno definitivamente acorta la esperanza de vida.
Frecuencia
La enfermedad de Batten y otras formas de ceroidolipofuscinosis neuronales son relativamente raras y se dan entre 2 a 4 de cada 100 mil nacimientos en los Estados Unidos. Estos trastornos parecen ser más comunes en Finlandia, Suecia, otras partes de Europa del Norte y en la provincia de Terranova en Canadá. Aunque las ceroidolipofuscinosis neuronales se clasifican como enfermedades raras, atacan a menudo a más de una persona en las familias que presentan los genes defectuosos.
Herencia
Las ceroidolipofuscinosis neuronales infantiles son trastornos recesivos autosómicos; es decir, ocurren solamente cuando un niño hereda dos copias del gen defectuoso, uno de cada padre. Cuando ambos padres poseen un gen defectuoso, cada uno de sus niños tiene una posibilidad en cuatro de padecer una ceroidolipofuscinosis neuronal. Al mismo tiempo, cada niño también tiene una posibilidad en dos de heredar solo una copia del gen defectuoso. A los individuos que poseen solamente un gen defectuoso se les conoce como portadores, lo que significa que ellos no desarrollan la enfermedad pero que pueden trasmitir el gen a sus propios niños. Debido a que se conocen cuáles son los genes mutados que están involucrados en ciertas formas de la enfermedad de Batten, la detección del portador es posible en algunos casos.
La ceroidolipofuscinosis neuronal del adulto puede ser heredada como un trastorno autosómico recesivo y, con menos frecuencia, como un trastorno autosómico dominante. En casos de herencia autosómica dominante, los individuos que heredan una sola copia del gen de la enfermedad la desarrollan. Por lo tanto, no hay portadores del gen que no estén afectados por la enfermedad.
Causa
Los síntomas de la enfermedad de Batten y de otras ceroidolipofuscinosis neuronales se asocian a una acumulación de sustancias llamadas lipofucinos (lipopigmentos) en los tejidos del cuerpo. Estos lipopigmentos se componen de grasas y de proteínas. Su nombre viene de la palabra técnica lipo, que es una abreviación de la palabra "lípido" o grasa y del término pigmento, denominado así porque las sustancias adquieren un color amarillo verdoso cuando se les visualiza con un microscopio de luz ultravioleta. Los lipopigmentos se acumulan en las células del cerebro y del ojo, así como en la piel, los músculos y muchos otros tejidos. En las células, estos pigmentos forman depósitos con formas particulares que pueden visualizarse con un microscopio electrónico. Algunos parecen medialunas, otros parecen huellas digitales. Los médicos buscan estos depósitos cuando examinan una muestra de piel para diagnosticar la enfermedad de Batten.
Los defectos bioquímicos que constituyen la base de varias ceroidolipofuscinosis neuronales se han descubierto recientemente. Se ha demostrado que una enzima llamada tioesterasa de proteína palmitóilica tiene una actividad insuficiente en la enfermedad de Batten infantil (esta condición se conoce actualmente como CLN1). En la forma preadolescente de esta enfermedad (CLN2), se ha detectado que la causa de esta condición es una deficiencia de una proteasa ácida, una enzima que hidroliza las proteínas. Se ha identificado un gen mutado en la enfermedad de Batten juvenil (CLN3), pero no se ha identificado la proteína correspondiente a este gen.
Diagnóstico
Debido a que la pérdida de la vista es a menudo un síntoma precoz, la enfermedad de Batten se puede detectar primero durante un examen de la vista. Un oftalmólogo puede detectar una pérdida de células en el ojo que ocurre en las tres formas infantiles de ceroidolipofuscinosis neuronales. Sin embargo, debido a que esta pérdida de células ocurre en otras enfermedades oftalmológicas, el trastorno no puede ser diagnosticado únicamente por este síntoma. A menudo, el oftalmólogo u otro médico especialista puede sospechar la presencia de una ceroidolipofuscinosis neuronal puede referir al niño a un neurólogo, un médico que se especializa en las enfermedades del cerebro y del sistema nervioso.
Para diagnosticar una ceroidolipofuscinosis neuronal, el neurólogo necesita el historial médico y varias pruebas de laboratorio del paciente. Las pruebas de diagnóstico utilizadas para detectar las ceroidolipofuscinosis neuronal incluyen:
- Análisis de sangre o de orina. Estas pruebas pueden detectar ciertas anormalidades que podrían indicar la presencia de la enfermedad de Batten. Por ejemplo, niveles elevados de una sustancia llamada dolicol que se encuentra en la orina de muchos pacientes que padecen de ceroidolipofuscinosis neuronales.
- Muestras de piel o de tejido. El médico puede examinar una muestra pequeña de tejido utilizando un microscopio electrónico. La ampliación de gran alcance del microscopio ayuda al médico a detectar depósitos típicos de las ceroidolipofuscinosis neuronales. Estos depósitos son comunes en las células de la piel, especialmente las de las glándulas sudoríparas (que producen el sudor).
- Electroencefalograma o EEG. Al realizar EEG se colocan parches especiales en el cuero cabelludo del paciente para registrar corrientes eléctricas en el cerebro. Esto ayuda a los médicos a examinar los patrones indicadores de la actividad eléctrica del cerebro que señalan si el paciente ha padecido convulsiones.
- Estudios eléctricos de los ojos. Estas pruebas, que incluyen respuestas de evocación visual y electroretinogramas, pueden detectar varios problemas del ojo frecuentes en las ceroidolipofuscinosis neuronales infantiles.
- Exploraciones del cerebro. Estas imágenes pueden ayudar a los médicos a detectar cambios en el aspecto del cerebro. Una técnica comúnmente utilizada en el procesamiento de imágenes es la tomografía computarizada o CT (por su sigla en inglés), que utiliza radiografías y una computadora para crear una imagen sofisticada de los tejidos y de las estructuras del cerebro. La tomografía computarizada puede revelar las áreas del cerebro que están sufriendo desgastes en pacientes que padecen ceroidolipofuscinosis neuronales. Otra técnica de procesamiento de imágenes que se está haciendo cada vez más común es el procesamiento de imágenes por resonancia magnética, o MRI (por su sigla en inglés). MRI utiliza una combinación de campos magnéticos y de ondas de radio, en vez de la radiación, para crear una imagen del cerebro.
- Medición de la actividad enzimática. La medición de la actividad tioesterasa de proteína palmitóilica ácida asociada al CLN1 y de la proteasa ácida asociada al CLN2 en los glóbulos blancos o en cultivos de fibroblastos de la piel se puede utilizar para confirmar estos diagnósticos.
- Análisis del ADN. Si se sabe que en la familia existe una mutación (alteración) del gen que produce la CLN3, se pueden utilizar análisis de ADN para confirmar el diagnóstico o realizar un diagnóstico prenatal de esta variación de la enfermedad de Batten. Cuando se conoce la mutación, el análisis de ADN se puede utilizar también para detectar los portadores no afectados de esta condición y poder realizar así un asesoramiento genético.
Tratamiento
Hasta ahora, no se conoce ningún tratamiento específico que pueda detener o revertir los síntomas de la enfermedad de Batten o de otras ceroidolipofuscinosis neuronales. Sin embargo, en algunos casos se pueden reducir o controlar las convulsiones con medicamentos anticonvulsivos y se pueden tratar otros problemas médicos apropiadamente a medida que se presentan. Al mismo tiempo, la terapia física y ocupacional puede ayudar a los pacientes a conservar el funcionamiento de su organismo por el mayor tiempo posible.
Algunos informes han señalado retardos de la enfermedad en niños que padecen de la enfermedad de Batten que fueron tratados con las vitaminas C y E y con dietas bajas en vitamina A. Sin embargo, estos tratamientos no previnieron que los pacientes murieran a causa de la enfermedad.
Ayudar y estimular a los pacientes puede contribuir a que ellos y sus familias puedan hacer frente a las situaciones graves de incapacidad y demencia causadas por las ceroidolipofuscinosis neuronales. A menudo, los grupos de ayuda permiten a niños, adultos y a las familias afectadas compartir preocupaciones y experiencias comunes.
Mientras tanto, los científicos prosiguen con la investigación médica que podría dar como resultado un tratamiento eficaz en el futuro.
Investigación
En el gobierno federal de los Estados Unidos, el punto focal que lleva a cabo la investigación sobre la enfermedad de Batten y otros trastornos neurogenéticos es el Instituto Nacional de Trastornos Neurológicos y Accidentes Cerebrovasculares (NINDS por su sigla en inglés). El NINDS, que forma parte de los Institutos Nacionales de la Salud, es responsable de apoyar y llevar a cabo las investigaciones sobre el cerebro y el sistema nervioso central. Gracias al trabajo de varios equipos científicos, la búsqueda de las causas genéticas de las ceroidolipofuscinosis neuronales está cobrando velocidad.
Otros investigadores también están trabajando en identificar qué sustancias constituyen los lipopigmentos. Aunque los científicos saben que los depósitos de los lipopigmentos contienen grasas y proteínas, no ha podido determinarse con exactitud la identidad de muchas de las moléculas de dichos depósitos. Los científicos han descubierto pistas potencialmente importantes. Por ejemplo, un científico patrocinado por el NINDS ha utilizado modelos animales de ceroidolipofuscinosis neuronales y ha descubierto que gran parte de este material acumulado es una proteína llamada sub-unidad c. Esta proteína se encuentra normalmente dentro de las mitocondrias de las células, estructuras pequeñas que producen la energía que las células necesitan para hacer su trabajo. Actualmente, los científicos están trabajando en entender qué papel puede desempeñar esta proteína en las ceroidolipofuscinosis neuronales, incluyendo cómo esta proteína termina encontrándose en la ubicación incorrecta y acumula células enfermas interiores. Otros investigadores también están examinando estos depósitos para identificar qué otras moléculas contienen.
Además, los científicos investigadores están trabajando con modelos animales de ceroidolipofuscinosis neuronales para entender y tratar mejor estos trastornos. Por ejemplo, un equipo de investigación está probando la utilidad del trasplante de médula en un modelo de ovejas, mientras que otros investigadores están trabajando en desarrollar modelos en ratas. Los modelos en ratas facilitarán que los científicos estudien la genética de estas enfermedades, puesto que las ratas se reproducen rápidamente y con gran frecuencia.
Referencias
- National Institute of Neurological Disorders and Stroke: publicado bajo dominio público.
NINDS (2003) "Enfermedad de Batten", National Institute of Neurological Disorders and Stroke: Office of Communications and Public Liaison; Publicado en noviembre de 2003 y revisado Revisado el 15 de julio de 2008; Publicación de NIH 04-2790; Bethesda, MD 20892;
Enlaces externos
¿Cómo puedo ayudar a la investigación?
El NINDS apoya a dos bancos humanos nacionales de especimenes cerebrales. Estos bancos proveen a investigadores alrededor del mundo tejido de pacientes de enfermedades neurológicas y psiquiátricas. Ambos bancos necesitan el tejido cerebral de pacientes que padecen la enfermedad de Batten para permitir que los científicos estudien más profundamente estos trastornos. Los posibles donantes o sus familias deben entrar en contacto con:
- Human Brain and Spinal Fluid Resource Center ; Neurology Research (127A) ; W. Los Angeles Healthcare Center ; 11301 Wilshire Blvd. Bldg. 212; Los Ángeles, CA 90073 ; 310-268-3536 ; 24-hour pager: 310-636-5199; Email: RMNbbank@ucla.edu ; http://www.loni.ucla.edu/~nnrsb/NNRSB
- Francine M. Benes, M.D., Ph.D., Director ; Harvard Brain Tissue Resource Center ; McLean Hospital ; 115 Mill Street ; Belmont, Massachusetts 02478; 800-BRAIN-BANK (800-272-4622) ; (617) 855-2400 ; http://www.brainbank.mclean.org
Existen dos organizaciones no financiadas por el NINDS que también proveen a los científicos investigadores tejido del sistema nervioso de pacientes con trastornos neurológicos. Los donantes interesados deben escribir o llamar a:
- National Disease Research Interchange ; 1628 JFK Blvd. ; 8 Penn Cntr. 8th floor ; Filadelfia, PA 19103 ; htor@ndri.com ; http://www.ndri.com
Tel: 215-557-7361 800-222-NDRI (6374) ; Fax: 215-557-7154
- Brain Endowment Bank ; Universidad de Miami; 1501 N.W. Ninth Avenue, #4013; Miami, FL 33136 ; Tel: 305-243-6219 800-UM-BRAIN (862-7246)
¿Dónde puedo encontrar más información?
Para obtener información adicional sobre los programas investigación del NINDS, contacte a la Unidad de Recursos Neurológicos y Red de Información del Instituto (BRAIN por su sigla en inglés) en:
- BRAIN; P.O. Box 5801; Bethesda, MD 20824; (800) 352-9424; http://www.ninds.nih.gov
Organizaciones
- Batten Disease Support and Research Association; 166 Humphries Drive; Reynoldsburg, OH 43068; bdsra1@bdsra.org
http://www.bdsra.org ; Tel: 800-448-4570 740-927-4298; Fax: 740-927-7683
- Children's Brain Disease Foundation [A Batten Disease Resource]; Parnassus Heights Medical Building, Suite 900
Suite 900; San Francisco, CA 94117 ; jrider6022@aol.com ; Tel: 415-665-3003; Fax: 415-665-3003
- Nathan's Battle Foundation [For Batten Disease Research]; 459 South State Road 135; Greenwood, IN 46142; pmilto@indy.net ;
http://www.nathansbattle.com ; Tel: 317-888-7396; Fax: 317-888-0504 lipidosislipidosis neuronales]]
Categorías:- Enfermedades hereditarias
- Enfermedades neurológicas
- Enfermedades metabólicas
- Tesaurismosis
- Trastornos generalizados del desarrollo
- Enfermedades degenerativas
- Enfermedades epónimas

Wikimedia foundation. 2010.