- Enfermedad de Pompe
-
Enfermedad de Pompe 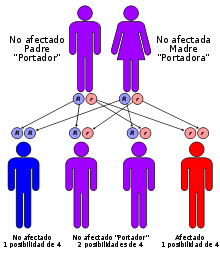
Clasificación y recursos externos CIE-10 E74.0 CIE-9 271.0 OMIM 232300 DiseasesDB 5296 PubMed Buscar en Medline mediante PubMed (en inglés) MeSH D006009  Aviso médico
Aviso médico La glucogenosis tipo II o enfermedad de Pompe, es una rara enfermedad de almacenamiento lisosómico hereditaria autosómica recesiva, causada por una disfunción de la enzima Glucosil Transferasa α(1→4) ácida lisosómica, también denominada maltasa ácida. Provoca una acumulación creciente de glucógeno en el lisosoma, que afecta, principalmente, al tejido muscular. En niños destaca por producir insuficiencia cardíaca al acumularse en el músculo cardíaco causando cardiomegalia.
Se estima que la incidencia de todos los subtipos clínicos es de uno por cada 40.000 nacimientos. La enfermedad de Pompe se da en todas las razas y, al ser una enfermedad autosómica recesiva, afecta por igual a hombres y mujeres. Se calcula que, tan sólo en los países desarrollados, puede haber entre 5.000 y 10.000 enfermos vivos. Se han detectado casos en distintas especies animales, incluyendo peces, aves y mamíferos.
De acuerdo con los datos de la Asociación Española de Enfermos de Glucogenosis (AEEG), en España la enfermedad presenta su mayor incidencia en las comunidades autónomas de Andalucía, Madrid y Murcia, siendo la provincia de Jaén el mayor foco aparente de esta patología, particularmente en sus variedades más graves.
Contenido
Causa de la enfermedad de Pompe
La enfermedad de Pompe es un error congénito del metabolismo del glucógeno que afecta al gen encargado de dar la orden de síntesis de la enzima alfa-(1,4)-glucosidasa en los lisosomas. Dicho gen (GAA) se encuentra localizado en el brazo largo del cromosoma 17 (17q). Dependiendo del tipo de mutación en el gen, existirá una deficiencia total o parcial de la actividad de la enzima en todas las células del organismo. Esta deficiencia puede tener consecuencias sobre diferentes tejidos, aunque el efecto más notable se produce en las células musculares, pues en ellas se acumula gran cantidad de glucógeno residual que es absorbido por los lisosomas para su transformación en glucosa. El depósito creciente de glucógeno en los lisosomas interfiere con la función celular y causa daños en las células.
Se han identificado cerca de 200 mutaciones del gen GAA que pueden encontrarse en la siguiente base de datos: Pompcenter.
Ver más información acerca de enfermedad de pompe en entorno medico
Subtipos clínicos
Existen tres variedades de la enfermedad de Pompe: la infantil, la juvenil y la adulta, definidas cada una de ella según la edad de aparición de los síntomas y la velocidad de progresión de la enfermedad, estando ambos parámetros determinados por el grado de actividad enzimática del paciente (inferior al 1% de los valores normales en la variedad infantil, entre el 1% y el 10% en la juvenil, y entre el 10% y el 20% en la adulta)
Tratamiento
Complicaciones cardíacas y respiratorias son tratados sintomáticamente. Terapias físicas y ocupacionales puede ser beneficiosa para algunos pacientes. Las alteraciones en la dieta pueden proporcionar mejoras temporales pero no alteraran el curso de la enfermedad. El asesoramiento genético puede proveer a las familias la información con respecto al riesgo en los embarazos futuros.
El 28 de abril de 2006 la Agencia de Medicamentos y Alimentos de los E.E.U.U. aprobó un uso de licencia biológico (BLA) para Myozyme (alglucosidase alfa, rhGAA),el primer tratamiento para los pacientes con la enfermedad de Pompe. Esto estaba basado en la terapia del reemplazo de una enzima, usando la alglucosidase alfa humana biológicamente activa, producida en células de los ovarios de los hámsters chinos. Myozime esta designada por el FDA como una droga huérfana, y fue aprobada en revisión de prioridad.
El FDA aprobó la administración de Myozime por infusión intravenosa de la solución. La seguridad y la eficacia de Myozyme fueron determinadas en dos ensayos clínicos separados en 39 pacientes con inicio infantil de la enfermedad de Pompe sus edades eran de un 1 mes a 3.5 años a la hora de la primera infusión. El tratamiento de Myozime prolonga claramente la supervivencia total. El diagnostico y tratamiento temprano conlleva a resultados mucho mejores. El tratamiento tiene efectos secundarios entre los cuales podemos encontrar fiebre,erupción cutánea y incremento del ritmo cardíaco, estas condiciones son usualmente manejables.
Myozime cuesta en promedio unos 300.000$ al año, y debe ser tomado por el paciente por el resto de su vida. Algunas aseguradoras estadounidenses se han negado a pagar por él. El 14 de Agosto de 2006 Health Canada aprobó Myozime para el tratamiento de la enfermedad de Pompe. El 14 de Junio de 2007 la revisión canadiense del medicamento común publicó sus recomendaciones con respecto a la financiación pública para la terapia de Myozyme. Su recomendación era proporcionar la financiación para tratar un subconjunto muy pequeño de los pacientes de Pompe (niños de menos de un año de edad con cardiomiopatía). La gran mayoría de países desarrollados está proporcionando el acceso a la terapia para todos los pacientes diagnosticados de Pompe. El 26 de mayo de 2010 fue aprobado Lumizyme por la FDA, una versión similar de Myozyme, para el tratamiento de la enfermedad de Pompe en inicios tardíos.
Véase también
- Medidas extraordinarias (película)
Enlaces externos
- Asociación Española de Enfermos de Glucogenosis (web) también en facebook AEEG
Categorías:- Hepatología
- Enfermedades metabólicas
- Enfermedades raras

Wikimedia foundation. 2010.