- Espectroscopia mediante resonancia magnética nuclear de proteínas
-
Espectroscopia mediante resonancia magnética nuclear de proteínas
 Introducción de la muestra en el espectrómetro RMN de alto campo magnético (800 MHz) del Pacific Northwest National Laboratory.
Introducción de la muestra en el espectrómetro RMN de alto campo magnético (800 MHz) del Pacific Northwest National Laboratory.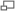
La espectroscopía mediante resonancia magnética nuclear de proteínas (usualmente llamada RMN de proteínas) es un campo de la biología estructural en el cual se utiliza espectroscopía RMN para obtener información sobre la estructura y dinámica de las proteínas. El campo fue desarrollado por Kurt Wüthrich, entre otros, quién compartió el premio Nobel de química en 2002. Las técnicas de RMN se utilizan en foma rutinaria en el ámbito académico y en la industria de biotecnología. La determinación de la estructura de las proteínas mediante espectroscopía RMN por lo general consiste en varias fases sucesivas, cada una de ellas utilizando un conjunto de técnicas altamente especializadas. La muestra es preparada, se asignan las resonancias, se generan las restricciones y se calcula y valida la estructura.
Contenido
Preparación de la muestra
Colección de datos
Asignación de resonancias
Para analizar los datos de resonancia magnética es nesario obtener la asignación de las resonancias de la proteína, es decir, descubrir qué desplazamiento químico corresponde a cada núcleo atómico. Para lograrlo, se han dearrollado distintos tipos de experimentos, dependiendo de si la proteína está isotópicamente etiquetada o no, pues muchos de los experimentos de asignación dependen del carbono-13 y el nitrógeno-15.
Generación de restricciones
Para realizar cálculos de estructura es necesario generar una serie de parámetros experimentalmente determinados.
Restricciones de distancia
Un "pico de cruce" en un experimento NOESY indica proximidad espacial entre los dos núcleos. Cada pico puede convertirse, por tanto, en una distancia máxima entre los núcleos, normalmente entre 1,8 y 6 angstroms. La intensidad del pico es proporcional a la distancia elevada a -6, de modo que la distancia se determina de acuerdo a la intensidad del pico.
La asignación de picos puede realizarse manualmente, pero resulta mucho más efectivo automatizar esta tarea mediante programas computacionales como CYANA[1] y ARIA[2]/CNS.
Restricciones de ángulos
Las restricciones en los ángulos de torsión de los enlaces químicos, normalmente los ángulos psi y phi pueden generarse de dos modos:
- Mediante la ecuación Karplus, para generar restricciones angulares a partir de las constantes de acoplamiento
- Mediante el uso de los desplazamientos químicos.
Ambos métodos utilizan el hecho de que la geometría alrededor del carbono alfa afecta a las constantes de acoplamiento y a los desplazamientos químicos, de modo que dados alguno de estos dos datos, pueden estimarse los ángulos de torsión.
Restricciones de orientación
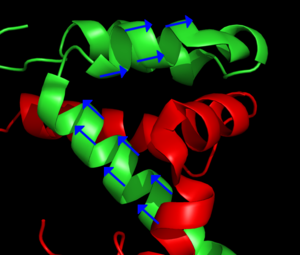 Las flechas azules representan la orientación del enlace N - H de enlaces peptídicos seleccionados. Mediante la orientación de un número suficiente de enlaces relativos al campo magnético externo, puede determinarse la estructura de la proteína. Tomado de PDB 1KBH.
Las flechas azules representan la orientación del enlace N - H de enlaces peptídicos seleccionados. Mediante la orientación de un número suficiente de enlaces relativos al campo magnético externo, puede determinarse la estructura de la proteína. Tomado de PDB 1KBH.Las moléculas del analito en una muestra pueden ordenarse parcialmente en relación al campo magnético del espectrómetro mediante la manipulación de las condiciones de la muestra. Las técnicas más comunes incluyen la adición de bacteriófagos o bicelas o la preparación de la muestra en un gel de poliacrilamida. Esto genera un entorno local que favorece ciertas orientaciones de moléculas no esféricas. Normalmente, en la RMN en solución el acoplamiento dipolar entre núcleos se promedia, debido al rápido tumbling de la molécula. El acoplamiento dipolar suele utilizarse en la RMN de estado sólido y proporciona información sobre la orientación relativa de los vectores de enlace en relación a un marco de referencia global. Típicamente, la orientación del vector N-H se determina en un experimento tipo HSQC. [3]
Intercambio hidrógeno-deuterio
Cálculo de la estructura
Archivo:Ensemble of NMR structures.jpgLa determinación de la estructura por RMN genera un conjunto de estructuras entre las cuales sólo unas pocas convergerán si los datos son suficientes para dictar un plegamiento específico. En estas estructures, es sólo el caso para una parte de la estructura. Obtenido de PDB 1SSU.Las restricciones determinadas experimentalmente pueden ser utilizadas como input para el proceso de cálculo de la estructura tridimensional. Los programas computacionales, como CYANA o XPLOR-NIH,[4], tratan de satisfacer tantas restricciones como sea posible, además de propiedades generales de las proteínas como la longitud de los enlaces y los ángulos. Los algoritmos convierten las restricciones experimentales y las propiedades generales de las proteínas en términos energéticos, y tratan de minimizar la energía. Este proceso resulta en un conjunto de estructuras que convergen si los datos son suficientes para dictar un plegamiento determinado.
Dinámica
La RMN puede ofrecer información sobre la dinámica de varias partes de la proteína, además de sobre la estructura. Esto implica medir tiempos de relajación como T1 y T2 para determinar parámetros de orden, tiempos de correlación y tasas de intercambio químico. La relajación (RMN) es una consecuencia de los campos magnéticos locales que fluctúan en una molécula, generados por movimientos moleculares. De este modo, las medidas de los tiempos de relajación pueden aportar información de los movimientos dentro de una molécula a nivel atómico. En los estudios RMN de dinámica molecular, el isótopo nitrógeno-15 es el núcleo preferido, porque sus tiempos de relajación son relativamente simples para relacionarlos con los movimientos moleculares que, sin embargo, requieren marcar isotópicamente la proteína. Los tiempos de relajación T1 y T2 pueden medirse utilizando varios tipos de experimentos basados en HSQC. Los tipos de movimientos que pueden ser detectados son aquellos que ocurren en una escala de tiempo de entre 10 picosegundos y 10 nanosegundos. Además, pueden estudiarse movimientos más lentos que tienen lugar en un rango temporal de entre 10 microsegundos y 100 milisegundos. No obstante, puesto que los átomos de nitrógeno se encuentran principalmente en el esqueleto de la proteína, los resultados reflejan principalmente los movimientos del esqueleto, que es la parte más rígida de la proteína. Por lo tanto, los resultados obtenidos por la medida de relajación del nitrógeno-15 pueden no ser representativos de la proteína entera. De ahí que, recientemente, se hayan desarrollado técnicas que utilizan medidas de relajación de carbono-13 y deuterio, lo que permite estudios sistemáticos de los movimientos de las cadenas laterales de aminoácidos.
Espectrometría RMN de proteínas grandes
Automatización del proceso
Véase también
- Cristalografía de rayos X
- Resonancia magnética nuclear
- Espectroscopia de resonancia magnética nuclear
Referencias
Citas
- ↑ Protein structure determination in solution by NMR spectroscopy Wuthrich K. J Biol Chem. 1990 December 25;265(36):22059-62
- ↑ Automated NMR structure calculation with CYANA. Guntert P. Methods Mol Biol. 2004;278:353-78.
- ↑ ARIA2: automated NOE assignment and data integration in NMR structure calculation. Rieping W, Habeck M, Bardiaux B, Bernard A, Malliavin TE, Nilges M. Bioinformatics 2007;23:381-382.
- ↑ An efficient 3D NMR technique for correlating the proton and 15N backbone amide resonances with the alpha-carbon of the preceding residue in uniformly 15N/13C enriched proteins. Bax A, Ikura M. J Biomol NMR. 1991 May;1(1):99-104.
- ↑ Residual dipolar couplings in protein structure determination. de Alba E, Tjandra N. Methods Mol Biol. 2004;278:89-106
- ↑ The Xplor-NIH NMR molecular structure determination package. Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. J Magn Reson. 2003 Jan;160(1):65-73
- ↑ Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Pervushin K, Riek R, Wider G, Wuthrich K. Proc Natl Acad Sci U S A. 1997 November 11;94(23):12366-71.
- ↑ Effect of deuteration on the amide proton relaxation rates in proteins. Heteronuclear NMR experiments on villin 14T. Markus MA, Dayie KT, Matsudaira P, Wagner G. J Magn Reson B. 1994 Oct;105(2):192-5
- ↑ NMR analysis of a 900K GroEL GroES complex. Fiaux J, Bertelsen EB, Horwich AL, Wuthrich K. Nature. 2002 July 11;418(6894):207-11.
- Guntert, P. See above
- Rieping, W. See above
- ↑ NMR data collection and analysis protocol for high-throughput protein structure determination. Liu G, Shen Y, Atreya HS, Parish D, Shao Y, Sukumaran DK, Xiao R, Yee A, Lemak A, Bhattacharya A, Acton TA, Arrowsmith CH, Montelione GT, Szyperski T. Proc Natl Acad Sci U S A. 2005 July 26;102(30):10487-92.
Literatura relacionada
- Gordon S. Rule, T. Kevin Hitchens (2006). "Fundamentals of Protein NMR Spectroscopy". Springer. ISBN 1-4020-3499-7. http://www.springer.com/1-4020-3499-7
- Quincy Teng, (2005). "Structural Biology, Practical NMR Applications, Springer, ISBN 0-387-24367-4
- John Cavanagh, Wayne J. Fairbrother, Arthur G. Palmer III, Nicholas J. Skelton, (1995). Protein NMR Spectroscopy: Principles and Practice. Academic Press. ISBN 0-12-164490-1.
- Kurt Wuthrich (1986) NMR of Proteins and Nucleic Acids . Wiley-Interscience. ISBN 0-471-82893-9
Enlaces relacionados
- Herramienta Didáctica para el Estudio de los Principios Físicos de la Imagen por Resonancia Magnética
- NOESY-Based Strategy for Assignments of Backbone and Side Chain Resonances of Large Proteins without Deuteration (a protocol)
- ProSA-web Servicio web para el reconocimiento de errores en la determinación de estructuras de proteínas
Categorías: Biofísica | Proteómica | Resonancia magnética nuclear | Métodos de proteína

Wikimedia foundation. 2010.