- Síndrome de Turcot
-
Síndrome de Turcot 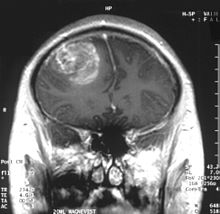
Imagen por resonancia magnética en corte coronal de la cabeza que demuestra la presencia de glioblastoma multiforme en el cerebro, uno de los tipos de tumores del sistema nervioso central que Jacques Turcot describió asociado a poliposis colorrectal.[1]Clasificación y recursos externos CIE-10 C71 C72 D12.6 CIE-9 191 192 211.3 CIE-O 8440/0 (poliposis adenomatosa), 9440/3 (glioblastoma) y 9470/3 (meduloblastoma). OMIM 276300 DiseasesDB 29793 Medline Buscar en Medline mediante PubMed (en inglés) Sinónimos "Síndrome de tumor cerebral y poliposis"; "Síndrome de glioma y poliposis".  Aviso médico
Aviso médicoEl Síndrome de Turcot, también denominado "Síndrome de tumor cerebral y poliposis" o "Síndrome de glioma y poliposis", es un trastorno hereditario caracterizado por el desarrollo de neoplasias malignas en el sistema nervioso central (por ejemplo, glioblastoma multiforme o meduloblastoma) asociadas a poliposis en el colon y en el recto, con pólipos que suelen ser el origen de cáncer colorrectal. Se han definido dos tipos de este síndrome,[2] cada uno con características clínicas diferentes, y se ha establecido que la causa de ambos es monogénica, es decir, causada por la mutación en un único gen, que es diferente para cada tipo del síndrome.
Contenido
Historia
El síndrome de Turcot fue descrito por primera vez en 1959 en el hospital Hôtel-Dieu de Québec, Canadá, por los doctores Jacqes Turcot, Jean Paul Després y François St. Pierre.[1] [2] Ellos reportaron dos casos del síndrome, en el cual dos hermanos (hombre y mujer) lo padecían. El hermano, de 15 años, consultó por diarrea y hematoquecia (deposiciones con sangre fresca). Le fueron hallados múltiples pólipos en el colon distal y en el recto, algunos de los cuales habían evolucionado a adenocarcinomas in situ. Le fue practicada una colectomía parcial y la resección en varias ocasiones de pólipos rectales. Fallece dos años después de la consulta inicial, y en la autopsia se halla destrucción completa de la médula espinal por causa de un meduloblastoma que la había infiltrado. La hermana, de 13 años, consulta por los mismos síntomas del hermano (diarrea y hematoquecia), y también le son encontrados múltiples pólipos en el colon distal y en el recto. Se le realizan dos colectomías para la estracción final de todo el colon, y le son destruidos periódicamente pólipos en el recto. Ocho años después de la consulta inicial, la paciente es hospitalizada por hallazgos compatibles con un tumor cerebral, falleciendo días después. La autopsia revela la presencia de un glioblastoma multiforme en el lóbulo frontal izquierdo (cerebro).
Clasificación
A medida que se han descrito casos del síndrome de Turcot, se ha hecho evidente que existen dos variantes de éste en cuanto son observadas diferencias en el patrón de herencia, las características clínicas y la etiología de cada uno. De esta manera, se tipifican dos tipos, el 1 y el 2, del síndrome de Turcot.
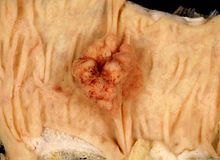 Esta foto muestra la mucosa del colon, con el eje longitudinal de éste en el eje horizontal de la foto. En el centro es visible un pólipo adenomatoso, una de las características síndrome de Turcot, además de los tumores del sistema nervioso central.
Esta foto muestra la mucosa del colon, con el eje longitudinal de éste en el eje horizontal de la foto. En el centro es visible un pólipo adenomatoso, una de las características síndrome de Turcot, además de los tumores del sistema nervioso central.
Síndrome de Turcot tipo 1
Este tipo del síndrome de Turcot se caracteriza por heredarse de forma autosómica reseciva y presentar como principal característica los gliomas (neoplasias de células gliales en el sistema nervioso central), mientras que la poliposis en el intestino grueso es secundaria. Así mismo, este subtipo del síndrome de Turcot se ha asociado con genes relacionados con la reparación de los errores de emparejamiento del ADN (o genes MMR, por la siglas en inglés de Mismatch Repair), como lo son los genes MSH2, MSH6, MLH1 y PMS2[2] . El carácter autosómico recesivo implica que los dos alelos del gen afectado estén mutados.
El síndrome de Turcot tipo 1 también se denomina "Síndrome de tumor cerebral y poliposis tipo 1", "Síndrome de glioma y poliposis tipo 1" o "Síndrome de cáncer por no reparación de los errores de emparejamiento" (en inglés Mismatch repair cancer syndrome[2] ), este último técnicamente más apropiado y adoptado por el proyecto Online Mendelian Inheritance in Man[1] (OMIM).
Síndrome de Turcot tipo 2
El síndrome de Turcot tipo 2 es considerado una variante de la poliposis adenomatosa familiar, dado que comparte con ésta el carácter hereditario autosómico dominante, además de poseer la misma etiología de las otras variantes de dicha enfermedad, a saber, mutaciones en el gen APC. Así mismo, a diferencia del tipo 1, el tipo 2 presenta como principal característica la poliposis intestinal de tipo adenomatoso con gran potencial de malignización, y como caracterítica secundaria los gliomas. Este subtipo del síndrome también se denomina "Síndrome de tumor cerebral y poliposis tipo 2" o "Síndrome de glioma y poliposis tipo 2".
Referencias
- ↑ a b Turcot J, Després JP, St.-Pierre F (September 1959). «Malignant tumors of the central nervous system associated with familial polyposis of the colon: report of two cases» (en inglés, PDF). Diseases of the Colon and the Rectum 2 (5): 465-468. doi:. PMID 13839882. https://tspace.library.utoronto.ca/bitstream/1807/17612/1/Turcot%20FAP%20glioblastoma.pdf. Consultado el 10 de octubre de 2011.
- ↑ a b c d Online Mendelian Inheritance in Man - OMIM (19 de julio de 2010). «Mismatch Repair Cancer Syndrome» (en inglés). Consultado el 7 de octubre de 2011.
Categorías:- Cáncer hereditario
- Enfermedades raras
- Síndromes
Wikimedia foundation. 2010.