- Poliposis adenomatosa familiar
-
Poliposis adenomatosa familiar 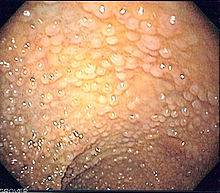
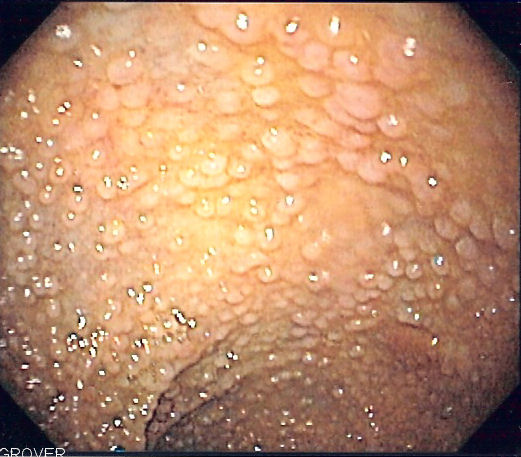
Endoscopia del colon sigmoide en paciente con Poliposis adenomatosa famliar que muestra múltiples pólipos.Clasificación y recursos externos CIE-10 D12.6 CIE-9 211.3 CIE-O 8140/0; 8220/3 OMIM 175100 DiseasesDB 4678 Medline Buscar en Medline mediante PubMed (en inglés) MedlinePlus 000266 eMedicine med/769 MeSH D011125  Aviso médico
Aviso médicoLa Poliposis adenomatosa familiar es una enfermedad hereditaria infrecuente que se incluye entre los Síndromes de poliposis intestinal[1] [2] , caracterizada por la aparición de gran número (más de 100 en la forma clásica) de pólipos del tipo adenomatoso (tumores benignos) en el colon y recto a partir de los 20 o 30 años. Dichos pólipos tienen una gran probabilidad de malignizarse a partir de los 30 años y transformarse en cáncer de colon.
Contenido
Variantes
Existen variantes de la Poliposis adenomatosa familiar, en base a las cualidades y cantidades de los pólipos, así como de otros fenómenos que pueden acompañar la poliposis[1] [2] . Aunque la mayoría de las variantes de la Poliposis adenomatosa familiar tienen la misma etiología, esta es, mutaciones en el gen APC, se han demostrado otras causas de síndromes hereditarios de poliposis adenomatosa[1] .
Se han establecido como variantes:
Poliposis adenomatosa familiar clásica
Caracterizada por tener como patrón de herencia el autosómico dominante, presentar múltiples pólipos adenomatosos intestinales (más de cien) con altísimo potencial de malignización localizados principalmente en colon y recto, además de la ausencia o baja intensidad de manifestaciones en otras ubicaciones corporales.
Poliposis adenomatosa familiar atenuada
Variante de la Poliposis adenomatosa familiar que a diferencia de la forma clásica, la cantidad de pólipos es menor de cien, manteniendo, sin embargo, un patrón de herencia autosómico dominante. Es un factor de riesgo de alta probabilidad para el desarrollo de cáncer colorrectal, así como factor de riesgo para otros tipos de cánceres.
Síndrome de Gardner
Caracterizado por múltiples pólipos en el colon que se asocian a[1] [3] :
-
- Adenomas periampollares (en referencia a la ampolla hepatopancreática)
- Quistes epidérmicos.
- Osteomas en la mandíbula, cráneo y huesos largos del organismo.
- Carcinoma papilar de tiroides
- Hepatoblastoma
- Tumores desmoides.
Se considera que el síndrome de Gardner no es más que una de las formas en que se puede manifestar la Poliposis adenomatosa familiar.
Síndrome de Turcot tipo 2
Véase también: Síndrome de TurcotEl síndrome de Turcot se caracteriza por tumores malignos del sistema nervioso central (i.e. glioblastoma multiforme y meduloblastoma) asociados a poliposis colónica. Se han descrito dos grupos dentro de este síndrome[4] :
-
- Tipo 2 (variante de la Poliposis adenomatosa familiar)
- Ciertos casos de Síndrome de Turcot se caracterizan etiológicamente por mutaciones en el gen APC, son de herencia autosómica dominante y la principal manifestación es la poliposis colónica con alto potencial de malignización, mientras que los tumores del sistema nervioso central son adicionales. Son estos casos del síndrome los que se consideran variantes de la Poliposis adenomatosa familiar.
- Tipo 1 (no considerado variante de la Poliposis adenomatosa familiar)
- En contraposición, otros casos de síndrome de Turcot son de herencia autosómica recesiva, la principal característica es la presencia de tumores en el sistema nervioso central y se han asociado con mutaciones en los genes MMR (por la siglas en inglés de Mismatch Repair).
Poliposis asociada a MUTYH
Denominada también Poliposis adenomatosa colorrectal autosómica recesiva, es una variante poco frecuente de la Poliposis adenomatosa familiar que a diferencia de otras, no es causada por mutaciones en el gen APC sino en el gen MUTYH, además de poseer un patrón de herencia autosómico recesivo.
Etiología
Está causado por una mutación en el gen APC situado en el cromosoma 5, en la zona de transición entre las bandas q21 y q22 de este[4] . Se transmite de padres a hijos según un patrón autosómico dominante, lo que significa que si alguno de los padres posee un gen afectado afectado, existe una probabilidad del 50% de que la enfermedad afecte a cualquiera de los hijos por término medio, independientemente del sexo de los mismos.
Cuadro clínico
Además de las manifestaciones en el colon, pueden aparecer pólipos en el estómago o duodeno, quistes dermoides, hipertrofia del epitelio pigmentario de la retina, alteraciones óseas y dentales.
Referencias
- ↑ a b c d Hsu, Evelyn K; Mamula, Petar; Ruchelli, Eduardo D (3 de enero de 2011). «Intestinal Polyposis Syndromes» (en inglés). Reference. Medscape. Consultado el 6 de octubre de 2011.
- ↑ a b Wehbi, Mohammad (1 de septiembre de 2011). «Familial Adenomatous Polyposis» (en inglés). References. Medscape. Consultado el 3 de octubre de 2011.
- ↑ Chimenos-Küstner, Eduardo; Pascual, Montserrat; Blanco, Ignacio; Finestres, Fernando (2005). «Poliposis familiar hereditaria y síndrome de Gardner: Aportación de la exploración odontoestomatológica a su diagnóstico y descripción de un caso» (PDF). Med Oral Patol Oral Cir Bucal 25 (10): 402-9. ISSN 1698-4447. PMID 16264375. http://www.medicinaoral.com/medoralfree01/v10i5/medoralv10i5p402.pdf. Consultado el 06-10-2011.
- ↑ a b Online Mendelian Inheritance in Man - OMIM (19 de julio de 2010). «Mismatch Repair Cancer Syndrome» (en inglés). Consultado el 7 de octubre de 2011.
Categorías:- Enfermedades hereditarias
- Enfermedades del aparato digestivo
- Tumores del aparato digestivo
- Cáncer hereditario
-
Wikimedia foundation. 2010.