- Distrofia miotónica de Steinert
-
Distrofia miotónica de Steinert
Distrofia miotónica de Steinert
Clasificación y recursos externos Aviso médico
Aviso médico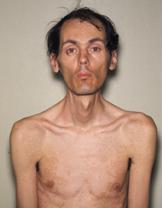
Paciente de 40 años de edad con distrofia miotónica presentando cataratas bilaterales y bloqueo cardíaco. CIE-10 G71 1 g70 OMIM 160900 Medline Buscar en Medline (en inglés) MedlinePlus D009223 Sinónimos Distrofia miotónica de tipo 1, Enfermedad de Curschmann-Steinert La distrofia miotónica, DM; también conocida como distrofia miotónica de tipo 1, DM1, es una enfermedad multisistémica crónica, de progresión lenta y de heredabilidad altamente variable que se puede manifestar en cualquier momento de la vida desde el nacimiento a la vejez. Se caracteriza por una reducción de la masa muscular (distrofia muscular), cataratas posteriores subcapsulares iridiscentes (opacidad del cristalino) defectos en la conducción del impulso cardiaco, cambios endocrinos y miotonía(dificultad para relajar un músculo). Es muy curioso que la edad de aparición, que es altamente variable, desciende con las sucesivas generaciones. Así pues la enfermedad muestra una edad de aparición cada vez menor, un fenómeno denominado anticipación. También hay dos clasificaciones de la DM, teniendo cada una de ellas diferentes síntomas asociados.
Contenido
Clasificación
La distrofia miotónica es la forma más común de aparición en el adulto de distrofia muscular y la segunda forma más habitual de enfermedad del músculo esquelético tras la Distrofia muscular de Duchenne. Actualmente se conocen dos tipos de DM de comienzo en la edad adulta: La distrofia miotónica tipo 1 (DM1), la conocida propiamente como enfermedad de Steinert, y la distrofia miotónica tipo 2 (DM2), denominada comúnmente como PROMM, o miopatía proximal miotónica. Ambas son identificables mediante análisis de ADN. La DM1 también tiene una forma congénita que puede afectar gravemente a bebes y tiene una forma de aparición en la infancia. Los investigadores sospechan que existen más formas de distrofia miotónica (DM3, DM4, DMX).
Diferencias entre DM1 y DM2
Aunque ambas enfermedades se consideran como afecciones degenerativas de desarrollo lento, la DM2 es generalmente más leve que la DM1. La forma congénita grave que afecta a los bebes en la DM1 no se ha encontrado en la DM2 y la literatura médica raramente publica apariciones juveniles de esta forma. La expansión de los trinucleótidos del DM2 es considerablemente más larga en la DM2 que en Dm1, variando de 75 a más de 11000 repeticiones. A diferencia de la DM1, el tamaño de las expansiones del trinucleótico en el ADN no parecen marcar ninguna diferencia en la edad de aparición o en la severidad de la enfermedad en la DM2. Parece que es menos significativo en el tipo 2 y las revisiones más habituales solo refieren que producen una ligera anticipación en éste último caso.
Epidemiología
Es la distrofia muscular más frecuente en el adulto. Su prevalencia se estima en 1/20000 habitantes.[1] . En la población canadiense de Saguenay-Lac St Jean estas cifras se elevan a 1/530 habitantes, siendo todos los afectados descendientes de una misma pareja de emigrados a Canadá en el S. XVII[2]
Etiología
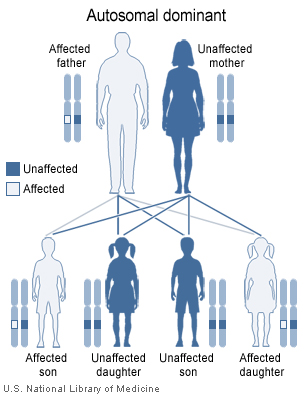 Distrofia miotónica con un patrón de dominancia autosómica
Distrofia miotónica con un patrón de dominancia autosómica
La DM es una enfermedad de transmisión genética donde está involucrado un patrón de dominancia autosómica, significando que hay un gen mutante de uno de los padres que resultará en esta condición. Hay un 50% de probabilidad de recibir DM del progenitor afectado. En la DM1, el gen afectado es el DMPK: proteína quinasa de distrofia miotónica, que codifica la kinasa miosina, expresada en los músculos esqueléticos. Este gen se localiza en el brazo largo del cromosoma 19. La DM2 es similarmente causada por un defecto del gen ZNF9 en el cromosoma 3 q21.
La DM es uno de los severos desórdenes de repetición de trinucleótidos. Ciertas áreas del ADN tienen secuencias repetidas de dos o tres nucleótidos. En DM1, hay una repetición triplete de citosina - timina - guanina (CTG) en el gen DMPK. El número de repeticiones varía grandemente de persona a persona, pero en promedio en un sujeto sano es entre 5 y 37. A veces cuando las secuencias repetitivas de ADN se replican en la división celular, la maquinaria celular agrega una copia extra de la repetición triple a la secuencia. Así hay 37 repeticiones triples en el gen DMPK, la secuencia comienza a ser inestable y los deslizamientos se hacen más probables de ocurrencia. La población afectada de DM1 tienen más de 50 y a veces tanto como 2000 repeticiones.
El resultado de esto es que el tamaño de repetición de un individuo con DM1 se hará cada vez más grande a medida que envejece. Así se explica el fenómeno de anticipación, así cada niño de un afectado adulto tendrá mayor expansión que su progenitor debido a deslizamientos durante la gametogénesis.
Los individuos con expansiones más grandes tienen más precoz el desorden y un más severo fenotipo. La expansión de repetición para DM2 es mucho más grande que en DM1, va de 75 a más de 11.000 repeticiones. Salvo DM1, el tamaño de la expansión del ADN repetido no aparece ser significativo en la edad de entrada o de severidad de la enfermedad en DM2. La anticipación aparece a ser menos significante en DM2 y más revisiones actuales reportan media anticipación como unm rasgo de DM2.
Cuadro Clínico
Las personas afectadas tienen una típica "cara en forma de hacha" producto de atrofia de los músculos temporales, masetero, y faciales.
Los hombres suelen presentar alopecia frontal. Los musculos del cuello y distales de los miembros se afectan de manera temprana.
Hay debilidad de los extensores de la muñeca, los dedos y musculos intrínsecos de la mano. Los musculos que permiten la dorsiflexion del tobillo están afectados provocando una caída del pie. Los musculos proximales suelen estar conservados, aunque muchos pacientes tienen afectación del cuadriceps. Hay afectación de musculos del paladar, farigeos y linguales que pueden provocar disartria, voz nasal y problemas en la deglucion. Muchos pacientes tienen debilidad de los músculos respiratorios, provocándoles una insuficiencia respiratoria.
La miotonia puede evidenciarse a partir de los 5 años de edad, pidiéndole al paciente que cierre la mano con fuerza, y luego la relaje, lo cual se produce de manera lenta.
Las manifestaciones cardiacas son comunes, fundamentalmente bloqueos AV de primer grado, entre otros trastornos eléctricos y prolapso mitral. Bloqueos completos pueden ocurrir ocasionando muerte subita del enfermo.
Diagnóstico
El diagnóstico de la DM1 suele producirse con facultativos con formación especializada en neurología y enfermedades neuromusculares del adulto. Sin la adecuada especialización y recursos médicos experimentados, es común que los pacientes acaben sin diagnóstico o con un diagnóstico erróneo. Aunque actualmente no existe cura para la DM y el tratamiento se basa en el alivio sintomático, se necesita en todo caso un diagnóstico preciso porque se pueden desarrollar otros problemas con el tiempo. Incluso los pacientes menos afectados pueden ser sometidos a chequeos rutinarios para complicaciones potencialmente fatales (p.ej. problemas con la conducción cardiaca, resistencia a la insulina, cataratas). Además el consejo genético debería estar disponible para todos los pacientes por el alto riesgo de transmisión.
Tratamiento
El tratamiento con quinina y procainamida pueden empeorar la conducción cardiaca, por lo que se recomienda sustituirlos por fenitoina. La colocación de un marcapasos debe ser considerada en enfermos que experimenten síncopes, o anormalidades severas de la conducción.
Referencias
- ↑ Prevalencia de la enfermedad de steinert en La Presse Médicale, Vol 36 - N° 6-C2 - Junio 2007
- ↑ Mathieu J, De Braekeleer M, Prevost C. Genealogical reconstruction of myotonic dystrophy in the Saguenay-Lac-Saint-Jean area (Quebec, Canada). Neurology 1990;40:839-84
Categorías: Enfermedades epónimas | Enfermedades genéticas | Neurología
Wikimedia foundation. 2010.