- Electrostática de macromoléculas
-
Electrostática de macromoléculas
La teoría electrostática clásica ha sido estudiada y aplicada en sistemas microscópicos de manera tal que se han obtenido resultados que comprueban su validez. Actualmente se tiene un fácil acceso a estudios de sistemas biológicos-moleculares de manera tal que se han realizado modelos que describen el comportamiento y las interacciones entre las estructuras moleculares. La electrostática de macromoléculas es la que trata de aplicar un modelo clásico, a sistemas como moléculas con una estructura compleja en la que se puede asumir ciertas consideraciones en su organización y aplicar los conceptos de la teoría electrostática clásica para describir sus interacciones.
El cálculo de los potenciales electrostáticos se puede efectuar haciendo una aplicación de la ecuación de Poisson en macromoléculas debido a que esta da una relación del potencial con la posición y la distribución de carga que lo genera. La respuesta del medio puede ser tratada por tres diferentes modelos para la reacción de las moléculas y los campos eléctricos:
- Polarizabilidad electrónica
- Dipolos permanentes
- Iones móviles
Contenido
Métodos
Cualquier cálculo, con independencia del modelo que se adopte, debe empezar con algunas consideraciones acerca de la estructura molecular, la distribución de carga y la respuesta ambiental del sistema de interés. La respuesta del ambiente es descrita por simulaciones y promedios por mecánica estadística, la medida de la constante dieléctrica o alguna representación de polarizabilidad como puntos de dipolos inducidos. La respuesta de los iones móviles es representada a través de la ecuación de Poisson-Boltzman o simulación de Monte-Carlo. Entonces, todos los modelos pueden ser reducidos a puntos o manchas cargadas y dipolos interactuando entre sí mismos y con el ambiente. En la práctica se trabaja solo con cargas, ya que la mayoría de tratamientos descompone los dipolos en pares de cargas parciales.
Para una reunión de cargas puntuales, qi, la energía total electrostática relativa con todas las cargas separadas infinitamente en el vacío es:
donde φi es el potencial experimentado por la carga qi. Teniendo en cuenta la interacción entre cargas y la interacción con el soluto:
donde
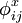 (x=c, e, s, m) es el potencial de carga i debido a la carga j. Los potenciales
(x=c, e, s, m) es el potencial de carga i debido a la carga j. Los potenciales 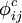 describen el potencial de Coulomb en el vacío. Entre todos los pares de cargas en la macromolécula;
describen el potencial de Coulomb en el vacío. Entre todos los pares de cargas en la macromolécula; 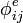 describe la interacción de las cargas con los electrones polarizables en la macromolécula. Los potenciales
describe la interacción de las cargas con los electrones polarizables en la macromolécula. Los potenciales 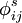 y
y 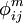 . Estos potenciales tienen contribuciones de la polarizabilidad electrónica , moléculas solventes iones móviles .
. Estos potenciales tienen contribuciones de la polarizabilidad electrónica , moléculas solventes iones móviles .Puesto que la interacción de Coulomb apantalla los efectos de los potenciales de interacción, en general todos estos efectos pueden ser combinados en una constante dieléctrica efectiva.
El método de las proteínas de Langevin
Esta aproximación trata de calcular la polarizabilidad electrónica por medio del modelo de dipolos inducidos, en donde cada átomo se asume con polarizabilidad uniforme representado como puntos dipolares que son situados en un enrejado, los cuales se comportan como la expresión de Langevin; no son tenidos en cuenta los puntos iónicos fuertes. Puesto que en este método se asumen puntos cargados tanto para el solvente y el soluto, y la ecuación de Langevin solo indica la dirección de los dipolos mas no la magnitud. A pesar de esto es un buen método para calcular las contribuciones de auto energía que hacen posible calcular las contribuciones electrostáticas para catálisis de enzimas en primera aproximación.
Solución analítica a la ecuación de Poisson-Boltzman
Para la solución analítica de la ecuación de Poisson-Boltzman (P-B) se considera que las macromoléculas tienen una constante dieléctrica uniforme baja y están sumergidas en un solvente con una constante dieléctrica igual a la del agua (aprox. 80). Además, se asume que la molécula tiene una forma geométrica conveniente y también el término del sinh(φ) en la ecuación de P-B en macromoléculas puede ser linealizado. Pese a estas consideraciones se han obtenido buenas aproximaciones, utilizadas en bastantes estudios como:
- en el estudio de las propiedades de membranas electrostáticas con la teoría de Gouy-Chapman se consideró una distribución de carga superficial sobre la membrana. Para cálculos sobre las propiedades eléctricas del ADN se le considera un cilindro uniformemente cargado, los resultados obtenidos por las consideraciones anteriores con la teoría de Manning´s en la que una parte de los aniones sobre el ADN se ligan a ella, haciendo un balance de las energías de dispersión entrópicas y de atracción entálpicas.
- Para un estudio aplicado a proteínas, la teoría de Tanford-Kirwood propone tratar a las proteínas como una esfera con una constante dieléctrica baja con cargas incrustadas, rodeada por un solvente con constante dieléctrica alta.
Solución numérica a la ecuación de Poisson-Boltzman
Los métodos numéricos para la solución de la ecuación de P-B son una herramienta fundamental porque en estos, a diferencia del método analítico, se puede considerar una forma compleja de la macromolécula reduciendo así el número de consideraciones y aproximaciones. Se ha utilizado para estudiar la estructura del ADN se desarrolló el método para obtener las distribuciones de iones, estos potenciales se comportan de acuerdo con la ley de Coulomb y se obtenía la distribución de cargas.
Método de las diferencias finitas para la ecuación de Poisson-Boltzman (FDPB)
El método FDPB considera a las moléculas y al solvente que los rodea ubicados en una rejilla cúbica, en cada punto de la rejilla se ubica una macromolécula o una molécula del solvente, a cada punto se le asigna una constante dieléctrica, una densidad de carga y la fuerza iónica que son parámetros de la ecuación de P-B, el hecho de que sea de diferencias finitas está en que las rejillas son pequeñas, se puede considerar la constante dieléctrica promediada sobre la molécula o no, puesto que el método no es ni microscópico ni macroscópico.
Véase también: Método de las diferencias finitasAplicaciones
Potenciales eléctricos alrededor de macromoléculas
Sobre la superficie de la macromolécula existen residuos cargados que generan potenciales eléctricos que se extienden varios angstroms dentro de la solución. Por ejemplo, en el DNA los potenciales producen gran concentración de asociación de iones, que afecta significativamente en un gran factor la conformación y las propiedades de los enlaces. El potencial eléctrico alrededor de las macromoléculas puede aumentar la razón de asociación del substrato, y el potencial eléctrico alrededor del DNA se ha creído responsable de la no secuencia específica de enlaces de proteínas y otras moléculas. El potencial eléctrico alrededor de ciertas proteínas puede ser el responsable del doblamiento del DNA.
El método FDPB es el camino disponible más efectivo para el cálculo de potenciales alrededor de macromoléculas. Simulaciones detalladas no son lo suficientemente amplias para ser significativo el efecto de la superficie o no incluyen los efectos de la fuerza iónica. El método de PDLD no es apropiado para el calculo de estos potenciales, en algunos casos no es razonable usar la ley de Coulomb con una constante dieléctrica de 80 o usar soluciones analíticas basadas en modelos simplificados de la macromolécula, además los efectos en los potenciales eléctricos producidos por el solvente pueden ser muy bajos si se utilizan métodos simplificados.
Hay estructuras que inducen gradientes de potencial grandes en sitios activos o potenciales exagerados en el interior de la solución. Los potenciales generados en el solvente muestran tener grandes efectos en la razón de asociación de moléculas cargadas. Este fenómeno recientemente ha sido estudiado de forma extensiva por el cálculo de Dinámica Browniana, el cual hace posible simulaciones de procesos de difusión. El método que ha sido más efectivo en el estudio de la asociación usa potenciales generados por cálculos de FDPB como bases para el cálculo de fuerzas usados en los procesos de simulación de la Dinámica Browniana.
Aplicaciones a proteínas Cu, Zn-superoxido dan un resultado satisfactorio del valor de la magnitud observada y la dependencia de la fuerza iónica de la razón constante para la difusión de los sitios activos, cuando se usaron modelos que utilizan potenciales simples de Coulomb los resultados difieren de los resultados experimentales.
Para el cálculo de potenciales alrededor de la macromolécula solo es necesario definir la superficie de Van der Waals y la distribución de carga y con solo una corrida del programa de FDPB se encuentran potenciales sobre un enrejado cúbico que pueden ser mostrados gráficamente o numéricamente. Por ejemplo, en el tRNA tiene una “ventana” de potencial negativo alrededor de la molécula, que fue encontrado en la región anticodon, esto sirve para reducir la interacción repulsiva con otras moléculas de cargada negativa y puede aumentar la interacción con el mRNA.
Energía de solvatación
El cálculo de energías de solvatación es importante porque las moléculas necesariamente cambian su estado en todos los procesos, como enlaces o arreglos conformacionales. Por ejemplo, cuando un substrato se enlaza a una proteína la interacción de ambas moléculas con el solvente cambia como resultado del proceso de enlace. En esta parte se trataran procesos en el que la molécula se transfiere de un medio a otro.
La fase gaseosa es un estado conveniente para el estudio de la solvatación, porque en este estado ninguna energía de interacción de soluto-solvente está presente. Entonces la energética de transferencia depende únicamente de las interacciones soluto-agua. Los procesos de solvatación implican huecos en la formación, cambian en las interacciones de van der Waals y en factores electrostáticos.
Las interacciones electrostáticas son factores dominantes en la solvatación de grupos cargados y generalmente también son significativos para grupos dipolares. Cada carga en una molécula crea un campo, lo que hace que el ambiente responda por polarización electrónica y reagrupamiento de carga, que resulta en una reacción potencial bajo la carga
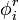 , la energía de una colección de cargas qien esta reacción potencial
, la energía de una colección de cargas qien esta reacción potencial 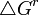 puede ser escrito como:
puede ser escrito como:El cambio en
sobre la transferencia de una carga o soluto polar de un solvente a otro define la componente electrostática de la energía de solvatación . φr contiene la contribución del solvente φs y iones móviles φm, si están presentes.La energía de solvatación puede ser calculada por un número de caminos. Métodos de perturbación de energía libre se han usado obteniendo la energía de solvatación vacío- agua con resultados bastante buenos. El método PDLD, produjo resultados cercanos a los experimentales. La ecuación de Poisson sirve como base para obtener las contribuciones electrostáticas para la energía de solvatación como otra opción.
Un ejemplo de la satisfactoria aplicación de la ecuación de Poisson es el modelo de Born para la solvatación de iones. Aunque cada Ion es tratado inicialmente como una esfera de uniformemente cargada, los iones también pueden ser tomados como puntos cargados en una esfera.
El cambio en la energía libre de solvatación [[\triangle\triangle G^{solv}]] en la transferencia de un ion de radio a y carga q de un medio de constante dieléctrica
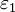 a un medio de constante dieléctrica
a un medio de constante dieléctrica 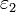 es:
es:El formalismo de la reacción potencial abre la posibilidad de usar la ecuación de Poisson para obtener la energía de solvatación para sistemas más complejos.
Catálisis
El potencial eléctrico de aminoácidos cargados en la superficie de proteínas puede influenciar la razón de asociación. Otro papel de la superficie cargada es el aumento de la razón catalítica de enzimas. EL método PDLD ha sido usado algunas veces para estudiar el papel de los aminoácidos cargados en sitios activos.
El método FDPB se ha utilizado para estudiar los efectos de la superficie cargada. En cálculos por este método son calculados los potenciales en sitios activos, generados por uno o más aminoácidos, y las energías electrostáticas son obtenidas por la multiplicación de estos potenciales por las cargas apropiadas considerando que se presentan en varios pasos en el mecanismo catalítico. Por ejemplo, cuando un protón es transferido la diferencia de potencial entre los dos sitios multiplicado por la carga del protón da la contribución energética en este paso.
Un número de cálculos, por el método de FDPB de potenciales eléctricos en sitio activos se han reportado, en estudios con actidin y papain campos electrostáticos debido a los aminoácidos cargados fueron evaluados y sugieren ser la base para las características reaccionantes de estas enzimas.
Para la trypsin, la estabilización electrostática de la transición de estado es importante, pero el efecto completo parece ser debido a grupos de puentes de hidrógeno y para la Asp-102 que es parte de sitios activos catalíticos, ambos cálculos PDLD y PDPB encontraron que la Asp-102 estabiliza la transición de estado en cerca de 4-5 Kcal/mol, en un excelente acercamiento a los resultados experimentales.
Fuerza en simulaciones dinámicas
Las simulaciones dinámicas moleculares requieren de las contribuciones electrostáticas para la fuerza de los átomos o grupos. El principal problema en estas simulaciones es la descripción del agua y del apantallamiento de los iones.
Una variedad de métodos han sido introducidos para imitar la respuesta dieléctrica del ambiente. Apantallamiento del solvente además del de los electrones y los dipolos en la macromolécula, debe ser tomada en cuenta una constante dieléctrica
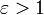 y varias funciones dieléctricas dependientes de la distancia.En simulaciones en el DNA, repulsiones entre fosfatos han sido minimizados por la omisión o reducción de la escala de sus cargas, se ha sugerido una aproximación alternativa en la determinación empírica basada en la derivación de la constante dieléctrica efectiva por cálculos por el método de FDPB.
y varias funciones dieléctricas dependientes de la distancia.En simulaciones en el DNA, repulsiones entre fosfatos han sido minimizados por la omisión o reducción de la escala de sus cargas, se ha sugerido una aproximación alternativa en la determinación empírica basada en la derivación de la constante dieléctrica efectiva por cálculos por el método de FDPB.Un problema adicional en muchas simulaciones en que la energía de solvatación no es tenida en cuenta. De manera similar, el factor intramolecular de los puentes de hidrógeno o formación de pares de iones, que ocurre a expensas de interacciones de magnitud comparable con el agua, es frecuentemente ignorado. En ausencia de una gran escala de simulaciones de solventes es importante el desarrollo de métodos para la medida del termino de solvatación, que puede ser un factor dominante en las energías conformacionales.
Bibliografía
- Antonio Blanco (2006). Qumica Biológica. Buenos Aries Editorial El Ateneo. ISBN 950-02-0422-3.
- Paul Lorrain (1970). Electromagnetic fields and waves. San Francisco : W.H. Freeman. ISBN 0-7167-0331-9.
Enlaces externos
- http://www.firp.ula.ve/cuadernos/S610A.pdf
- http://bacterio.uc3m.es/docencia/doctorado/sistemas_complejos/biofisica/biophys10.pdf
Categoría: Biofísica
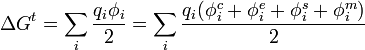
![\Delta G^{t} =\sum_{i} \frac{q_{i}[\phi_{ii}^{e}+\phi_{ii}^{s}+\phi_{ii}^{m}+\sum (\phi_{ij}^{c}+\phi_{ij}^{e}+\phi_{ij}^{s}+\phi_{ij}^{m})]}{2}](/pictures/eswiki/49/1bbbd9cc18e613629ee1463c12afbf99.png)
![\triangle
G^{r}=\sum_{i}\frac{q_{i}}{2}[\phi^{e}_{i}+\phi^{s}_{i}+\phi^{m}_{i}]=\sum_{i}q_{i}\phi_{i}/2](/pictures/eswiki/51/307afb2c527a69af4fb045f04e9cea8e.png)
![\triangle\triangle G^{solv}= \frac{q
\triangle\phi^{r}}{2}=\frac{q^{2}[\frac{1}{\varepsilon_{2}}-\frac{1}{\varepsilon_{1}}]}{2a}](/pictures/eswiki/52/4ce4c5a7a4c08d6f9466240530e70eb1.png)
Wikimedia foundation. 2010.