- Esclerosis tuberosa
-
Esclerosis tuberosa
 Entrada da Associação Brasileira de Esclerose Tuberosa, en Belo Horizonte.
Entrada da Associação Brasileira de Esclerose Tuberosa, en Belo Horizonte.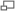
¿Qué es la esclerosis tuberosa?
La esclerosis tuberosa es una enfermedad hereditaria autosómica dominante con penetrancia incompleta, poco frecuente, que produce la formación de masas anormales (tumores no cancerosos) en algunos órganos del cuerpo, como pueden ser: la retina, la piel, los pulmones, los riñones y el corazón. Generalmente también suele afectar al Sistema Nervioso Central (la médula espinal y el cerebro)
Esta enfermedad entra dentro de un grupo de enfermedades llamadas síndromes neurocutáneos.
Esta enfermedad posee una variedad de nombres como son:
- Síndrome de Bourneville Pringle
- Esclerosis Tuberosa y Facomatosis
- Tuberoesclerosis
- Epiloia
Las lesiones cerebrales de la enfermedad, fueron descritas, por primera vez en el año 1862, por Recklinghausen. Bourneville, años más tarde, hizo publicas las manifestaciones anatomo-clínicas. El nombre esclerosis tuberosa se debe a los crecimientos producidos en el cerebro, en forma de raíz, que se van calcificando con la edad y se vuelven duros.
Etiología de la enfermedad
La esclerosis tuberosa es causada por mutaciones en dos genes (TSC1 y TSC2). Si se afecta uno de los genes puede ocurrir la enfermedad. El gen TSC1, mencionado anteriormente, se encuentra en el cromosoma 9 y da lugar a una proteína llamada hamartina. A diferencia del gen TSC2, que se encuentra situado en el cromosoma 16 y causa la proteína llamada tuberina. El gen TSC1 fue hallado en el 1997 y el gen TSC2 se descubrió en el año 1993. Los científicos opinan que estas proteínas ( hamartina y tuberina) intervienen como supresores del crecimiento del tumor, regular la diferenciación celular y los procesos de proliferación. En estos procesos las células nerviosas se parten para dar lugar a las nuevas generaciones de células y se obtienen características individuales.
 Esclerosis tuberosa.
Esclerosis tuberosa.Datos importantes
Los síntomas de la esclerosis tuberosa pueden presentarse en el momento de nacer. Aunque en algunas personas el avance de los síntomas pueden presentarse más tardes. Existe variabilidad en el grado de la enfermedad, es decir, algunos pacientes presentan una forma leve de la enfermedad, otros pueden presentar discapacidades severas. En casos excepcionales, las masas anormales pueden poner en peligro la vida No se necesita a los dos padres para transmitirse la mutación, con un solo miembro es suficiente para que el niño consiga la enfermedad. Aún así, en la mayoría de los pacientes con Esclerosis Tuberosa, se produce por nuevas mutaciones, por lo que generalmente no existe un antecedente familiar de la enfermedad, por lo que la enfermedad se obtiene a través de un proceso llamado mosaicismo gonadal (la mutación afecta a una parte de los gametos: óvulos o espermatozoides). En los últimos años, se ha detectado que el gen de la esclerosis tuberosa está enlazado al lugar de grupo sanguíneo ABO y al encogen c-abl (situados, los dos, en el brazo largo del cromosoma 9 (9 q 34)). Se necesitan más estudios para confirmar más locus genéticos, algunos ya evidenciados: 11q 14-23, 16q13.3.
Epidemiología
La Esclerosis tuberosa se da en personas de diferentes grupos étnicos y de ambos sexos. A nivel mundial, se afirma que hay cerca de 1 a 2 millones de individuos, y se cree que incide en 1 de cada 6000 recién nacidos. En EEUU, existirían entre 25 000 y 40 000 casos. La incidencia se ha calculado en menos de 1 caso por 100.000 persona/año.
Sintomatología
Manifestaciones cutáneas y mucosas
Manchas hipocrómicas o acrómicas
- Se trata de unas manchas que aparecen en la piel, en forma de hoja lanceolada o en hoja de fresno.
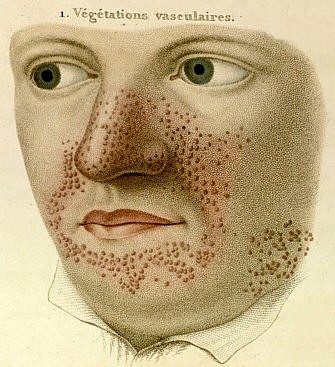
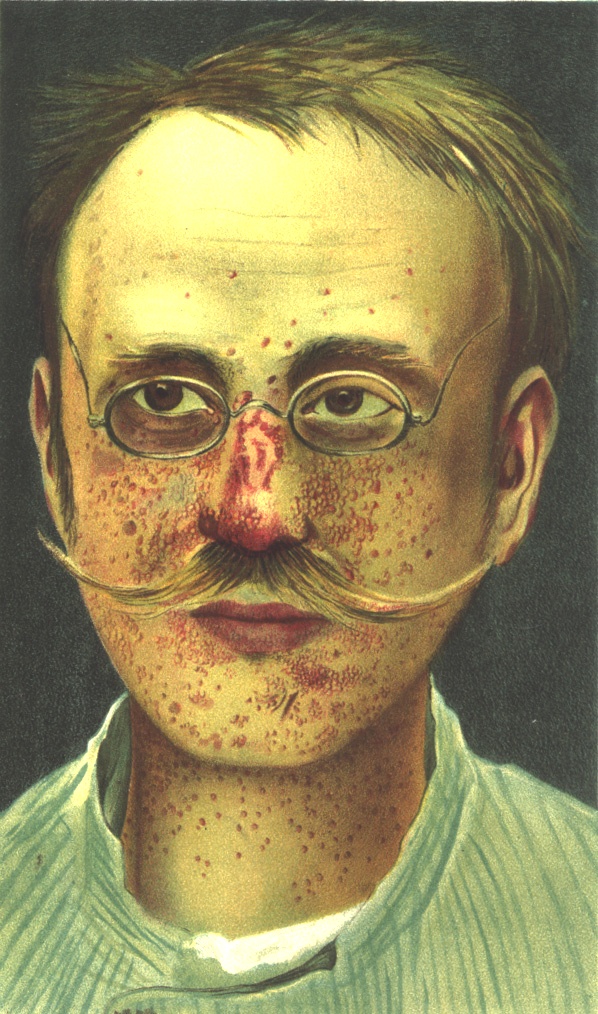
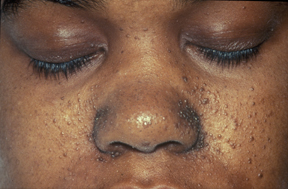
 Angiofibromas.
Angiofibromas.Los angiofibromas
- Son tumores redondeados de color rojizo que nacen de la dermis protruyendo sobre la epidermis, con un tamaño que oscila entre una cabeza de alfiler a un guisante, suelen aparecer en torno al mentón, mejillas y nariz en forma de alas de mariposa (surcos nasogenianos). Otra localización característica de los angiofibromas son subungueales o periungueales en las manos y los pies.
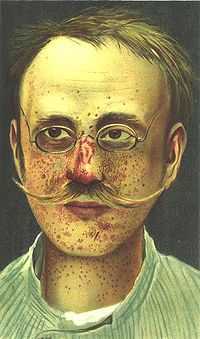
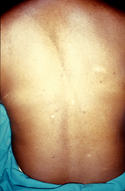
 Piel de naranja.
Piel de naranja.Piel de naranja
- Con una textura irregular como la naranja, generalmente en el dorsal o lumbar.
Manifestaciones neurológicas
Crisis epilépticas
- Alrededor del segundo año de vida en un 80-90% de los casos. Si se aparece tempranamente, suele cursarse en forma de sindrome de West o de Lennox-Gastaut.
Trastornos
- Trastornos mentales, trastornos del comportamiento o alteraciones psicóticas, pueden asociarse a la esclerosis tuberosa.
Otras manifestaciones
- Pueden ser las derivadas del aumento de la presión intracraneal (trastorno del sueño, convulsiones, cefaleas, etc.)
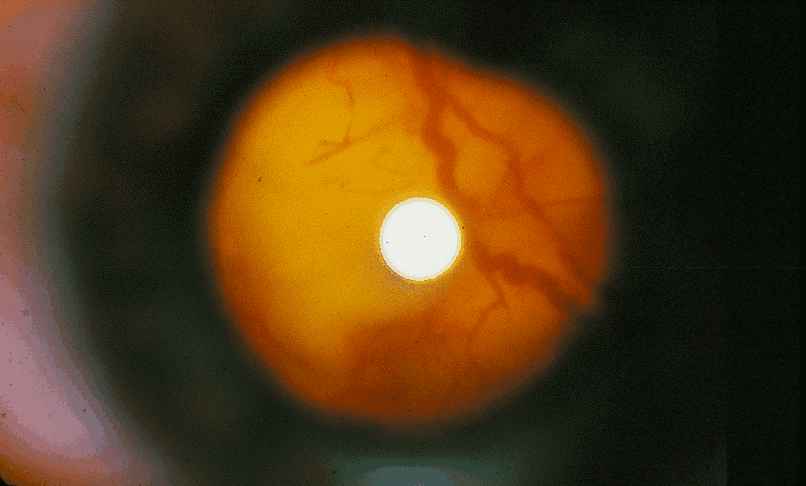 Hamartomas retinianos.
Hamartomas retinianos.Manifestaciones oftalmológicas
Hamartomas retinianos
Lesiones viscerales
Renales
- Angiomiolipomas, tumores benignos asociados a quistes.
Cardíacas
- Rabdomioma, no suele dar sintomatología.
Pulmonares
- Poliquistosis, suele ser la más común.
Vasculares
- Displasias. Algunas de éstas pueden ser aneurismas cerebrales.
Lesiones esqueléticas
Las lesiones oseas
- Suelen ser geodas de 1 a 3mm, seudoquísticas, metacarpianas y metartasianas o falángicas, o bien zonas de hiperostosis.
Manifestaciones orales
Placas fibrosas
- Ubicadas en la zona de las encías, labios y lengua.
Alteraciones dentales
- Hipoplasia del esmalte en forma de hoyo.
Lesiones mucosas
- Nódulos mucosos constituyen la manifestación más frecuente de éstas. Su coloración es rojo-amarillento o del color de la mucosa, localizados preferentemente en la porción anterior de la encía. En las encías; hiperplasia gingival aunque en mayor parte debida a tratamiento anticonvulsionante y mala higiene oral, más que a la enfermedad en sí.
Lesiones esqueléticas
- Paladar hundido, el labio fisurado y la hiperostosis.
Otras
- Tumor odontogénico calcificado, fibroma desmoplástico, hemangiomas mucosos y/o intraóseos, mixoma odontogénico, asimetría facial, úvula bífida, retraso de la erupción y diastemas, entre otras.
Tratamiento
Debido a la gran variedad de síntomas, y del amplio espectro que éstos pueden presentar, no existe un tratamiento específico para esta enfermedad. Así que el tratamiento se basa en tratar cada síntoma que presente la persona afectada.
Síntomas cutáneos
Angiofibromas
- Son adenomas sebáceos que se pueden eliminar con láser. También puede reiterarse su uso con las sucesivas recurrencias
Síntomas neurológicos
Crisis epilépticas
- Se utilizarán medicamentos antiepilépticos según prescriba el médico a fin de intentar controlar las crisis (ácido valproico, carbamazepina, etc…) aunque ello no garantiza el control de éstas.
Retraso mental
- Se puede necesitar la ayuda de educación especial dependiendo del grado de retraso.
Síntomas viscerales y esqueléticos
La actuación dependerá de la gravedad, localización y sintomatología de la lesión. Así, por ejemplo, el rabdomiomas no suelen dar síntomas y suelen desparecer con la pubertad, así que no es necesaria su extirpación. Otras afectaciones viscerales, como los quistes renales o pulmonares, aneurismas, tumores u otros requerirán tratamiento específico definido.
Evolución
La evolución de la esclerosis tuberosa tiene una tendencia de progreso conforme el afectado va avanzando en edad, al igual que van aumentando las alteraciones existentes. Según el órgano afectado del individuo, tendrá una evolución u otra y la edad de fallecimiento va en relación con el tamaño de los tumores. El pulmón, el riñón y el sistema nervioso central son los que determinan el pronóstico. Desde la aparición de la enfermedad, se tienen que tomar una serie de tratamientos de estimulación precoz (psicomotricidad, fisioterapia, logopedia…) resaltar que a lo largo de su vida necesitará apoyo en lecto-escritura y practicar natación para fortalecer los músculos por la hipotonía que producen las crisis epilépticas. En las diversas patologías, a parte de las crisis epilépticas, la más grave es el problema de conducta, por ello también es necesario apoyo psicológico para la orientación de sus familiares.
A lo largo de la evolución se realizarán una serie de intervenciones, electroencefalograma si hay crisis epilépticas, su periodicidad dependerá del grado de las crisis. TAC craneal cada cinco años, para un correcto control de los nódulos subependimarios y de su localización en relación al agujero de Monro. RM cerebral, en caso de que se plantee la exéresis quirúrgica de algún túber cerebral cortical, pues esta exploración define mejor las estructuras cerebrales que el TAC. Y por último, se hará psicometría y cuantificación del coeficiente intelectual, especialmente en niños con problemas escolares o en el momento de comenzar la escuela, para situarles en el nivel educativo adecuado.
Es preciso efectuar una vigilancia con práctica de controles periódicos, para detectar precozmente la aparición de complicaciones tumorales. Se ha señalado que la edad media del fallecimiento de estos pacientes se situaba en torno a los 24 años (Webb y Cols, 1996) pero otros estudios (Jancar, 1996) indican que su longevidad se ha incrementado en los últimos tiempos.
Prevención
El tratamiento más efectivo de esta entidad es su prevención.
En caso de antecedentes familiares se recomienda el consejo genético. Si uno de los padres se encuentra afectado, la posibilidad de transmitir la enfermedad se estima en el 50%. se deberia advertir a la pareja del riesgo que conlleva la procreacion mas no desaconsejarla, debido a que esto solo es decisión de la pareja.
Existe también la disponibilidad de diagnóstico prenatal de mutaciones conocidas en el ADN, pero ya que la enfermedad aparece frecuentemente como mutaciones nuevas, raramente se puede prevenir.
Preguntas frecuentes
¿La Esclerosis Tuberosa está catalogada como una enfermedad rara?
Si, ya que es muy poca conocida y tiene una reducida frecuencia en la población. Esta infrecuencia conlleva numerosas consecuencias adversas, tanto a nivel médico como social. Esta enfermedad pone en riesgo la vida de los enfermos ya que ha sido poco estudiada. Al ser tan poco conocida, su diagnóstico es tardío y los tratamientos ausentes en la mayoría de los casos. La ausencia de un tratamiento efectivo se debe a la escasez de investigación y a la ausencia de rentabilidad comercial de medicamentos destinados a un pequeño número de pacientes.
¿Qué causa la ET?
La ET es causada por defectos o mutaciones en dos genes, TSC1 y TSC2. Solamente uno de los genes necesita ser afectado para que ocurra el CET. El gen TSC1, descubierto en 1997, se encuentra en el cromosoma 9 y produce una proteína llamada hamartina. El gen TSC2, descubierto en 1993, se encuentra en el cromosoma 16 y produce la proteína llamada tuberina. Los científicos creen que estas proteínas actúan como supresores del crecimiento del tumor, agentes que regulan los procesos de proliferación y diferenciación celular, en los cuales las células nerviosas se dividen para formar las nuevas generaciones de células y adquirir características individuales.
¿Es hereditario el CET?
Cualquier hijo de una persona con E.T. tiene un 50 % de probabilidades (1 de cada 2) de heredar el gen. Pero incluso si buscamos hasta la última de las características posibles en los hijos de personas con E.T. encontramos menos del porcentaje previsto del 50 % con el gen. Ello se debe a su penetrancia reducida, es decir, que no todos los portadores del gen de la E.T. mostrarán signos de ello. Cuando se da en el caso más extremo se denomina no-penetrancia, y cuando, por ejemplo, un abuelo y un nieto tienen la E.T., pero el escalón intermedio (padre/madre) no muestra signos de ello, a eso se le llama " saltarse una generación”.
¿Qué investigación se están realizando?
Dentro del gobierno federal, el patrocinador principal de la investigación sobre el CET es el Instituto Nacional de Desórdenes Neurológicos y Accidentes Cerebrovasculares (NINDS). El NINDS es responsable de apoyar y de conducir la investigación sobre el cerebro y el sistema nervioso central. NINDS conduce la investigación en sus laboratorios en el NIH y también apoya estudios de investigación a través de patrocinios a las instituciones médicas importantes en todo el país.
 Dr. Laughlin Dawes.
Dr. Laughlin Dawes.Los científicos que estudian el CET buscan aumentar nuestra comprensión sobre el trastorno al aprender más sobre los genes TSC1 y TSC2 que pueden causar el trastorno y la función de las proteínas producidas por estos genes, la tuberina y la hamartina. Los científicos esperan que el conocimiento aportado por la investigación actual mejore las pruebas genéticas del CET y genere nuevas alternativas de tratamiento, métodos de prevención, y, en última instancia, la cura de este trastorno.
Otro estudio se enfoca en dos trastornos cerebrales importantes, el autismo y la epilepsia, que ocurre en niños que padecen de CET. La información obtenida en este estudio podría conducir a una mejor comprensión de los tres trastornos, así como la implementación de nuevos tratamientos y drogas. Otros científicos están intentando determinar qué causa los tumores de la piel en individuos que padecen de CET y en encontrar la base molecular de estos tumores. Los resultados de este estudio podrían ser sumamente útiles para ayudar a obtener nueva información sobre la genética del CET.
Una persona tiene ataques y tres puntos blancos (máculas hipopigmentadas). Su médico le aconsejó que se hiciera unas pruebas genéticas y resultaron negativas (no se identificaron cambios) ¿Qué significa esto?
Es posible que no tenga ET y solo tenga dos características de esta enfermedad, o puede padecerla y que tenga un caso leve de ET.
¿Cuántas personas tienen ET?
Un cálculo reciente afirma que 1 de cada 6.000 bebes nacidos padecerá esta enfermedad. Casi un millón de personas en el mundo la tiene y aproximadamente 50.000 en los Estados Unidos la padecen, pero también existen muchos casos en los que la enfermedad no se diagnostica debido a los leves síntomas o por la oscuridad de la enfermedad.
Padeciendo ET, ¿Cómo pueden verse tantos órganos afectados?
Porque ambos genes (el TSC1 Y TSC2) controlan el crecimiento celular del cuerpo, juegan un papel fundamental en el desarrollo fetal temprano del cerebro y de la piel. Por ello, cuando uno de los genes es defectuoso, el crecimiento no se inhibe y da como resultado ET.
¿Son cancerosos los tumores?
No, (excepto un 2% que pueden desarrollar tumores renales cancerosos) pero pueden provocar problemas, ya sea bloqueando los flujos de líquido cefalorraquídeo, con la aparición de tumores en el cerebro, con tumores en el corazón bloqueando el flujo sanguíneo y pueden provocar arritmias graves o lo tumores del riñón que pueden crecer demasiado y hacerse con la función normal lo que conlleva a una insuficiencia renal.
¿Cuál es la expectativa normal de una persona con ET?
Puede tener complicaciones en órganos como el riñón o el cerebro, lo que conlleva a serias dificultades incluso a la muerte si no recibe el tratamiento adecuado, por ello debe de someterse a pruebas médicas a lo largo de su vida para detectar posibles complicaciones.
Ya que no existe cura aún, ¿Qué puede hacerse?
Someterse a la cirugía para extirpar tumores o para detener el crecimiento tumoral, esto está ayudando a preservar la función de órganos afectados.
Por otro lado, para ayudar a controlar la epilepsia se están realizando pruebas para señalar con exactitud la localización precisa de convulsiones cerebrales.
¿Dónde puedo encontrar más información?
En la alianza de Esclerosis Tuberosa (Tuberous Sclerosis Alliance), puede estar informado de los avances que se realizan haciéndose miembro, así como explicaciones de su diagnostico, pruebas genéticas y cuidados y opciones de tratamiento.
- www.tsalliance.org
Para obtener información adicional sobre los programas investigación del Instituto Nacional de Desórdenes Neurológicos y Accidentes Cerebrovasculares ( NINDS), contacte a la Unidad de Recursos Neurológicos y Red de Información del Instituto BRAIN en: BRAIN P.O. Box 5801 Bethesda, MD 20824 (800) 352-9424 http://www.ninds.nih.gov
La información también está disponible de las organizaciones siguientes:Tuberous Sclerosis Alliance 801 Roeder Road Suite 750 Silver Spring, MD 20910-4467 info@tsalliance.org Esta dirección de correo electrónico está siendo protegida de \"spam bots\", necesita habilitar Javascript para poder verla. http://www.tsalliance.org**---Tel: 301-562-9890 800-225-6872 Fax: 301-562-9870
Epilepsy Foundation 4351 Garden City Drive Suite 500 Landover, MD 20785-7223 postmaster@efa.org Esta dirección de correo electrónico está siendo protegida de \"spam bots\", necesita habilitar Javascript para poder verla. http://www.epilepsyfoundation.org**---Tel: 301-459-3700 800-EFA-1000 (332-1000) Fax: 301-577-2684National Organization for Rare Disorders (NORD) P.O. Box 1968 (55 Kenosia Avenue) Danbury, CT 06813-1968 orphan@rarediseases.org Esta dirección de correo electrónico está siendo protegida de \"spam bots\", necesita habilitar Javascript para poder verla. http://www.rarediseases.org**---Tel: 203-744-0100 Voice Mail 800-999-NORD (6673) Fax: 203-798-2291
Bibliografía
Síndromes neurocutáneos en la infancia, Rafael Palencia Luaces, Valladolid: Secretario de Publicaciones e intercambio Científico, universidad de Valladolid, 1998, Capitulo 2 (pág.: 25-33)
Enlaces externos
Atención Multidisplinar a un Usuario con Esclerosis Tuberosa
Manifestaciones Cutáneas de la Esclerosis Tuberosa
Código: CIE-9-MC: 759.5
Categoría: Enfermedades genéticas

Wikimedia foundation. 2010.