- Síndrome de West
-
Síndrome de West Clasificación y recursos externos CIE-10 G.40X CIE-9 345.6 DiseasesDB 6788 eMedicine neuro/171 MeSH D013036 Sinónimos Síndrome de los espasmos infantiles
Encefalopatía epiléptica
Tic de Salaam
Espasmos en flexión
Lighting spells Aviso médico
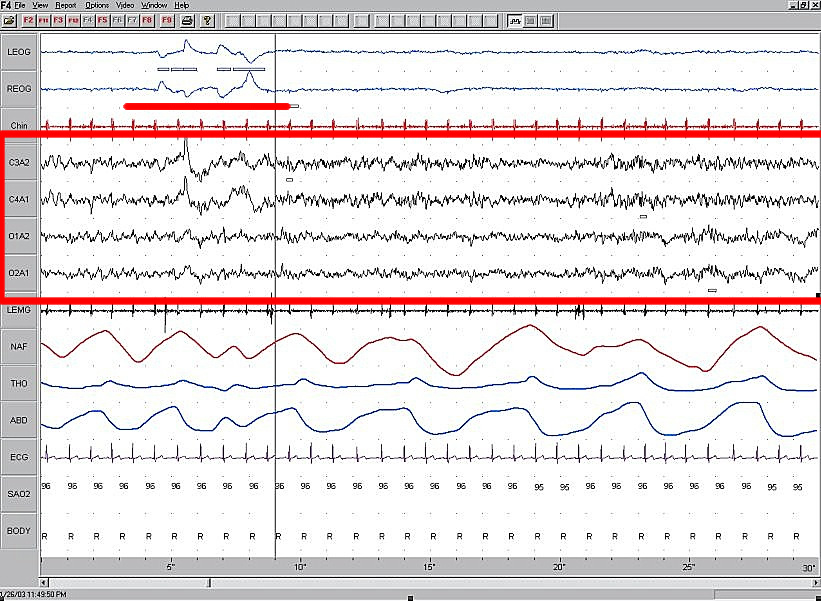
Aviso médico 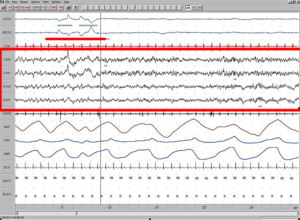 Electroencefalograma durante la fase REM del sueño. El diagnóstico de síndrome de West se realiza mediante la detección de hipsarritmias en el EEG, desapareciendo estas durante esta fase del sueño.
Electroencefalograma durante la fase REM del sueño. El diagnóstico de síndrome de West se realiza mediante la detección de hipsarritmias en el EEG, desapareciendo estas durante esta fase del sueño.
El síndrome de West (SW) o síndrome de los espasmos infantiles es una encefalopatía (alteración cerebral) epiléptica de la infancia, grave y poco frecuente, que debe su nombre a William James West (1793-1848), médico inglés que describió por primera vez el cuadro (presente en su propio hijo) en un artículo publicado por The Lancet en 1841.[1] Se caracteriza típicamente por tres hallazgos: espasmos epilépticos, retraso del desarrollo psicomotor y electroencefalograma con un trazado característico de hipsarritmia, aunque uno de los tres puede no aparecer.
Los niños con SW suelen manifestar la enfermedad entre los 3 y 6 meses de edad, aunque en ocasiones esto ocurre hasta los dos años. El SW siempre genera algún grado de retraso global en el desarrollo infantil y, a pesar de que el conocimiento sobre él ha mejorado considerablemente, todavía hay casos en los que no se diagnostica a tiempo, ante todo cuando los síntomas son leves (las convulsiones se pueden confundir con cólicos o dolor abdominal) o debido a la falta de experiencia por parte del pediatra.[2]
Contenido
Etimología y clasificación
El grupo de trabajo para la Clasificación y Terminología de la Liga Internacional contra la Epilepsia (ILAE) clasifica al SW, según su etiología, en sintomático y criptogénico. Se denomina SW sintomático al cuadro debido a una o varias lesiones estructurales cerebrales identificables, mientras que se reserva el término criptogénico para los casos en los que se supone dicha lesión pero no se consigue evidenciar o localizar. La ILAE no admite la existencia de casos idiopáticos (sin causa, y por lo tanto, sin lesión estructural), aunque varios autores han publicado algún caso que incluyen en esta categoría.[3] El SW sintomático es el más frecuente,[4] ya que la medicina moderna consigue encontrar en muchos casos la lesión estructural causante del cuadro. Las causas pueden ser prenatales (las más frecuentes), perinatales o postnatales. Otra clasificación muy empleada es la que habla de síndrome de West primario (el que aparece antes de los 3 primeros meses de vida), secundario (a partir de los 7-8 meses) y tardío (a partir de los dos años, siendo el primero de mejor pronóstico.[5]
Causas prenatales
- La más frecuente (30%) es la displasia cerebral. Dentro de esta categoría se incluyen la esclerosis tuberosa, la neurofibromatosis, el síndrome de Sturge-Weber o la microcefalia congénita. También se relaciona con el síndrome del nevus lineal sebáceo, la hemangiomatosis neonatal, el síndrome de Aicardi,[6] la holoprosencefalia o la esquizencefalia.
- Algunos trastornos cromosómicos también pueden ser causa prenatal del SW: El síndrome de Down, síndrome de Miller-Dieker, la duplicación del brazo corto del cromosoma 18 o la del 15.
- Infecciones como el citomegalovirus, herpes simple, rubéola, sífilis o toxoplasmosis, cuando afectan al feto pueden ser causa de SW.
- Trastornos metabólicos: fenilcetonuria, hiperglucemia, hiperornitinemia, síndrome de Leigh, deficiencia de piruvato-carboxilasa, deficiencia de piruvato deshidrogenasa, enfermedad de Krabbe, adrenoleucodistrofia neonatal, leucodistrofia leucocromática, encefalopatía por glicina o deficiencia de biotinidasa.
- Síndromes congénitos: Síndrome de Sjögren, síndrome de Smith-Lemli-Optiz o la enfermedad de Fahr.
- La hipoxia o la isquemia de causa prenatal (poroencefalia, hidranencefalia, leucomalacia periventricular...) son causantes, en ocasiones de la aparición del síndrome.
Causas perinatales
- Se definen como causas perinatales aquellas que tienen lugar entre la semana 28 del embarazo y la primera semana de vida tras el nacimiento. Se incluyen aquí la necrosis neural, el status marmoratus, la leucomalacia periventricular, la poroencefalia, o la encefalomalacia multiquística.
Causas postnatales
- Infecciones: meningitis bacteriana, absceso cerebral, meningoencefalitis vírica (sarampión, varicela,herpes simple, enterovirus, adenovirus, citomegalovirus o virus de Epstein-Barr).
- Hemorragias o traumatismos con consecuencia de hemorragia subdural o subaracnoidea.
Fisiopatología
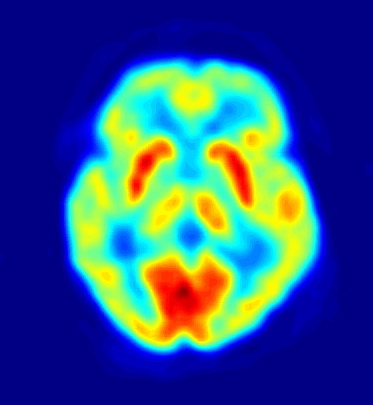
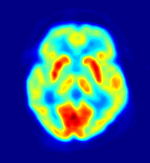 TEP. Las áreas en rojo indican alta actividad cerebral.
TEP. Las áreas en rojo indican alta actividad cerebral.Existen varias teorías que tratan de explicar el mecanismo de aparición del síndrome. La edad de aparición sugiere la implicación de fenómenos de inmadurez cerebral en la base del fenómeno. La edad típica de aparición coincide con la época de formación de dendritas y de mielinización de los axones neuronales, lo que parece avalar esa teoría. Diversos autores[7] postulan que un desequilibrio en la producción de neurotransmisores del tallo cerebral podría originar la hipsarritmia y los espasmos epilépticos (por aumento de los sistemas serotoninérgico o adrenérgico, o por inhibición del sistema colinérgico). Esta teoría se sustenta en la disminución de la duración de la fase REM del sueño en estos pacientes, que coincide con fases de disminución del patrón de hipsarritmias y con una menor frecuencia de aparición de espasmos. Varios estudios con tomografía por emisión de positrones y de flujo sanguíneo cerebral apoyan la influencia de estructuras o señales anómalas corticales en el desarrollo del cuadro. Por último algunos estudios apuntan a una relación entre el SW y alteraciones del sistema inmunitario: los pacientes presentan con mayor frecuencia que la población general el subgrupo de proteínas de antígeno HLA DRW52, y parece existir alguna alteración en las citocinas (niveles séricos elevados de la interleucina–2, factor de necrosis tumoral alfa e interferón alfa).
Epidemiología
La incidencia (frecuencia de aparición) de este síndrome es de 1/4000 a 6000 nacidos vivos, con predomino en varones (3:2). No existe una clara asociación familiar (excepto en la variedad relacionada con la esclerosis tuberosa), ni siquiera con otros cuadros epilépticos. En la mayoría de los casos (45 de 50) las crisis se inician entre el tercero y el duodécimo mes de vida. Con menos frecuencia aparecen en los primeros dos meses de vida, o entre los dos y los cuatro años. Este síndrome aparece en un 1-5% de los niños con síndrome de Down, siendo este un grupo de buena respuesta al tratamiento antiepiléptico. En estos niños los hallazgos del EEG son más simétricos y con menos anomalías que los que padecen el síndrome de West sin la trisomía del par 21. Se aducen razones genéticas para estas diferencias pero no se conocen los mecanismos exactos que las determinan.
Cuadro clínico
Espasmos epilépticos
Son contracciones bruscas, bilaterales y normalmente simétricas de la musculatura del cuello, tronco y extremidades, que se suelen acompañar de pérdida de conciencia. Pueden ser espasmos en flexión (cabeceo o encogimiento de hombros), en extensión (opistótonos), o mixtos.[8] Pueden aparecer diferentes tipos de espasmo en el mismo paciente o durante una crisis, aunque en general se consideran de peor pronóstico las crisis en las que predominan los espasmos asimétricos. Los espasmos rara vez se presentan aislados: suelen ocurrir en salvas (típicamente al despertarse, o antes de dormirse) y son muy poco frecuentes durante el sueño. A veces los espasmos se acompañan de síntomas vasovagales (sudoración, enrojecimiento facial, midriasis, ...) Se consideran "gatillos" o desencadenantes de una salva, el hambre, la excitación, una temperatura elevada o estímulos táctiles o sonoros.
Retraso psicomotor
Es común, incluso antes de la aparición de los espasmos, la detección de un grado variable de retraso psicomotor. Se evidencia, a edades tan tempranas, con signos como la pérdida de capacidad de seguimiento visual, disminución el reflejo de prensión, hipotonía muscular, hemiplejía (alteración simétrica de la movilidad en dos extremidades) o tetraplejía (en cuatro).
Alteraciones del EEG
Los hallazgos electroencefalográficos más específicos del SW son el enlentecimiento y la desorganización de la actividad eléctrica cerebral, en forma de trazado caótico con mezcla de puntas y ondas lentas independientes. A este patrón característico se le denomina hipsarritmia.
Diagnóstico y diagnóstico diferencial
Las manifestaciones clínicas suelen ofrecer una importante pista diagnóstica en la mayoría de los casos. De cualquier manera cualquier niño con manifestaciones epileptiformes debe realizarse un EEG, siendo esta la prueba diagnóstica esencial para la confirmación del síndrome, con el patognomónico hallazgo de hipsarritmia. Otros cuadros con los que se puede confundir son:
- Cólico del lactante
- Mioclonía benigna de la infancia temprana
- Reflujo gastroesofágico
- Epilepsia mioclónica del lactante
- Encefalopatía mioclónica precoz
- Síndrome de Ohtahara
Pronóstico
Este síndrome tiene, en general, mal pronóstico. El 90% de los casos presentan un retraso psicomotor importante, con limitaciones motoras y rasgos de personalidad autista. Tiene una mortalidad del 5%, y casi la mitad de los casos pueden desarrollar otros síndromes epileptiformes, como el síndrome de Lennox-Gastaut. Son de mejor pronóstico las variedades idiopáticas o criptogenéticas, y peor en los casos más sintomáticos. El grupo de niños con West y Down es de mejor pronóstico, como se comentó más arriba.
Tratamiento
Fármacos
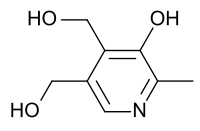 Piridoxina (Vitamina B6).
Piridoxina (Vitamina B6).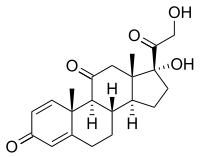 Prednisona.
Prednisona.- Anticomiciales (fenobarbital...) y sedantes: Durante las salvas de espasmos se emplean antiepilépticos o benzodiacepinas para el control de las mismas.
- Piridoxina: Es de primera elección, ya que este suplemento vitamínico soluciona aquellos casos en los que la causa es un déficit de vitamina B6.
- Hormona adrenocorticotropa (ACTH): Existen múltiples pautas de administración. Presenta una tasa de respuesta similar a los corticoides, pero debe valorarse su empleo debido a la toxicidad inducida que puede provocar (mortalidad atribuible del 5%, por hemorragias secundarias a hipertensión, o infecciones).
- Ácido valproico: Es un buen controlador de las crisis hasta en la mitad de los casos. Las dosis empleadas varían mucho (desde 40 mg/kg/día hasta 150 mg/kg/día). Tiene un efecto preventivo en el desarrollo del retraso psicomotor y trastornos de la conducta.
- Vigabatrina: De eficacia similar a la ACTH, pero con menos efectos secundarios, aunque se ha relacionado con trastornos visuales.
- Corticoides (Prednisona): Eficaz en el tratamiento de los espasmos, aunque presenta efectos secundarios como hipertensión, hiperglucemia, aumento de peso, irritabilidad...
- Topiramato: Fármaco antiepiléptico de amplio espectro que ha demostrado tasas de respuesta altas y buena tolerancia cuando se introduce progresivamente.
Cirugía
En los casos en los que no existe respuesta al tratamiento o este resulta contraindicado de manera absoluta se plantea la posibilidad de un abordaje quirúrgico para extirpar la zona de lesión cerebral. Suele ser una técnica eficaz en la resolución de las crisis, aunque su eficacia en el desarrollo psicomotor es más controvertida.
Referencias
- ↑ West WJ. On a particular form of infantile convulsions. Lancet 1841; 1: 724-5.
- ↑ Síndrome de West, epilepsia infantil de difícil control. Entrevista en saludymedicinas.com.mx con la Dra. Graciela Olmos García del Alba. http://www.saludymedicinas.com.mx/nota.asp?id=2201
- ↑ Dulac, Vigevano, Ohtahara y colaboradores
- ↑ Caraballo R, Cersósimo R, Galicchio S, Fejerman N. Epilepsias en el primer año de vida. Rev Neurol 1997; 25: 1521-4
- ↑ Caraballo R, Cersósimo R, Arroyo H, Fejerman N. Síndrome de West sintomático: asociaciones etiológicas particulares con respuesta inesperada al tratamiento. Rev Neurol 1998; 26: 372-5.
- ↑ Bour F, Chiron C, Dulac O, Plouin P. Caractères electrocliniques des crises dans le syndrome d’Aicardi. Rev Electroencephalogr Neurophysiol Clin 1986; 16: 341-53.
- ↑ Hrachovy et col.
- ↑ Vázquez HJ. Epilepsia en flexión generalizada. Arch Argent Pediatria1951; 35: 111-41.
Bibliografía
- West WJ. On a particular form of infantile convulsions. Lancet 1841.
- Vázquez HJ. Epilepsia en flexión generalizada. Arch Argent Pediatria.
- Caraballo R, Cersósimo R, Galicchio S, Fejerman N. Epilepsias en el primer año de vida. Rev Neurol 1997.
- Caraballo R, Cersósimo R, Arroyo H, Fejerman N. Síndrome de West sintomático: asociaciones etiológicas particulares con respuesta inesperada al tratamiento. Rev Neurol 1998.
Enlaces externos
Categorías:- Síndromes
- Enfermedades epónimas
- Epilepsia



Wikimedia foundation. 2010.