- Huella peptídica
-
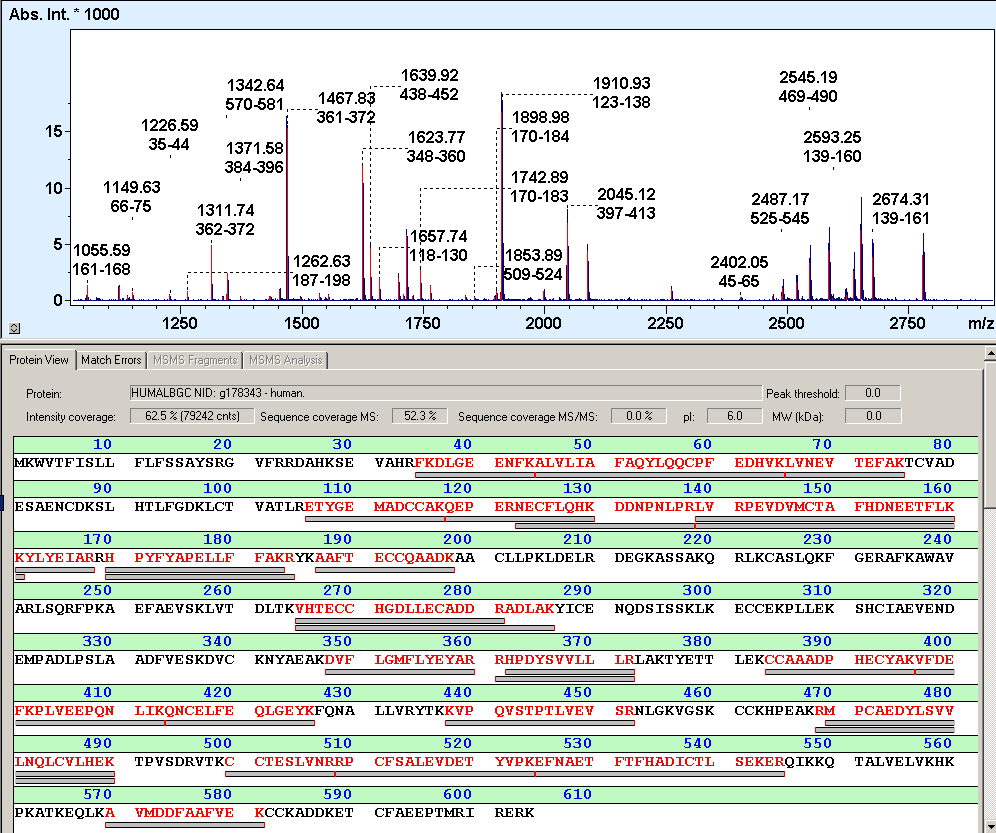
 Secuenciación de una proteína mediante huella peptídica.
Secuenciación de una proteína mediante huella peptídica.
La huella peptídica o PMF (del inglés Peptide mass fingerprinting), es una técnica analítica de identificación de proteínas desarrollada en 1993 por varios grupos de forma independiente.[1] [2] [3] [4] [5] En este método, la proteína desconocida en estudio es hidrolizada ( mediante proteasas específicas de secuencia, generalmente la tripsina) en pequeños péptidos cuyas masas absolutas pueden determinarse mediante un espectrómetro de masas acoplado al detector adecuado, como el MALDI-TOF o el ESI-TOF.[6] Las masas obtenidas son comparadas con una base de datos biológica de proteínas cuya secuencia se conoce o bien de información genómica, esto es, mediante un enfoque in silico. Para este menester se emplean herramientas de software capaces de traducir las secuencias nucleotídicas del genoma, depositadas en la base de datos, a secuencias de aminoácidos (esto es, los componentes de las proteínas); luego, se corta teóricamente la secuencia de la cadena polipeptídica y se calculan las masas absolutas de los péptidos obtenidos. La ulterior comparación entre huella de tamaños de péptidos obtenida con las depositadas en la base de datos permite asociar estadísticamente la proteína desconocida con la más semejante de la base de datos (asociació que puede ser de identidad). La mayoría de las bases de datos asumen que la proteína está constituida de un sólo péptido, lo que supone una desventaja.[7]
Para el uso de la técnica de la huella peptídica debe disponerse de la proteína pura, o, de existir mezclas de dos o tres, debe acoplarse un MS/MS adicional. Como paso preparativo, el aislamiento de la proteína de interés suele realizarse mediante electroforesis en gel bidimensional o mediante electroforesis SDS-PAGE. Los análisis adicionales mediante MS/MS pueden ser directos, generalmente mediante MALDI-TOF/TOF o nanoLC-ESI-MS/MS análisis de los elementos recortados del gel de electroforesis bidimensional.[7] [8] [9]
Referencias
- ↑ Pappin DJ, Hojrup P, Bleasby AJ (1993). «Rapid identification of proteins by peptide-mass fingerprinting». Curr. Biol. 3 (6): pp. 327–32. doi:. PMID 15335725.
- ↑ Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C (1993). «Identifying proteins from two-dimensional gels by molecular mass searching of peptide fragments in protein sequence databases». Proc. Natl. Acad. Sci. U.S.A. 90 (11): pp. 5011–5. doi:. PMID 8506346.
- ↑ Mann M, Højrup P, Roepstorff P (1993). «Use of mass spectrometric molecular weight information to identify proteins in sequence databases». Biol. Mass Spectrom. 22 (6): pp. 338–45. doi:. PMID 8329463.
- ↑ James P, Quadroni M, Carafoli E, Gonnet G (1993). «Protein identification by mass profile fingerprinting». Biochem. Biophys. Res. Commun. 195 (1): pp. 58–64. doi:. PMID 8363627.
- ↑ Yates JR, Speicher S, Griffin PR, Hunkapiller T (1993). «Peptide mass maps: a highly informative approach to protein identification». Anal. Biochem. 214 (2): pp. 397–408. doi:. PMID 8109726.
- ↑ Clauser KR, Baker P, Burlingame AL (1999). «Role of accurate mass measurement (+/- 10 ppm) in protein identification strategies employing MS or MS/MS and database searching». Anal. Chem. 71 (14): pp. 2871–82. doi:. PMID 10424174.
- ↑ a b Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, Mortensen P, Shevchenko A, Boucherie H, Mann M (1996). «Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels». Proc. Natl. Acad. Sci. U.S.A. 93 (25): pp. 14440–5. doi:. PMID 8962070.
- ↑ Wang W, Sun J, Nimtz M, Deckwer WD, Zeng AP (2003). «Protein identification from two-dimensional gel electrophoresis analysis of Klebsiella pneumoniae by combined use of mass spectrometry data and raw genome sequences». Proteome Science 1 (1): pp. 6. doi:. PMID 14653859.
- ↑ Hufnagel P, Rabus R (2006). «Mass spectrometric identification of proteins in complex post-genomic projects. Soluble proteins of the metabolically versatile, denitrifying 'Aromatoleum' sp. strain EbN1». J. Mol. Microbiol. Biotechnol. 11 (1-2): pp. 53–81. doi:. PMID 16825790.
Categorías:- Métodos de proteína
- Espectrometría de masas
Wikimedia foundation. 2010.