- SDS-PAGE
-
SDS-PAGE
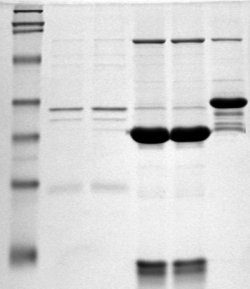
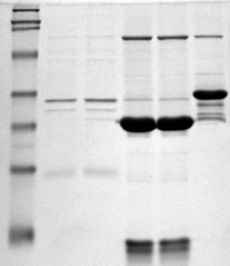 Imagen de SDS-PAGE. El marcador molecular corre en la calle de la izquierda.
Imagen de SDS-PAGE. El marcador molecular corre en la calle de la izquierda.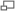
SDS-PAGE es el acrónimo en inglés de sodium dodecyl sulfate polyacrylamide gel electrophoresis (electroforesis en gel de poliacrilamida con dodecilsulfato sódico). Es una técnica ampliamente utilizada en bioquímica, genética, biología molecular y ciencia forense para separar las proteínas de acuerdo a su movilidad electroforética (en función de la longitud de la cadena polipeptídica, masa molecular, modificaciones postraduccionales y otros factores). Gracias al SDS la mayoría de proteínas adquieren una relación carga/masa idéntica, por lo que se obtiene un fraccionamiento que obedece únicamente a la longitud de la cadena. Es el método de electroforesis empleado con mayor profusión para analizar proteínas.
Contenido
Procedimiento
La disolución de proteínas que se van a analizar se mezcla en primer lugar con SDS, un detergente aniónico que desnaturaliza las proteínas, eliminando sus estructuras secundaria y terciaria (pero sin alterar los enlaces disulfuro y además confiere una carga negativa a cada proteína en proporción a su masa.[1] [2] [3] Sin SDS, las distintas proteínas que tienen masas moleculares similares migran de forma diferente debido a diferencias en la proporción carga/masa, ya que cada proteína tiene un punto isoeléctrico distinto. Si la electroforesis se realiza así, sin añadir SDS, se habla de PAGE nativa. El problema se resuelve añadiendo SDS, puesto que al unirse y desplegar la proteína, le proporciona una carga negativa casi uniforme a lo largo de la longitud de la cadena polipéptidica.
El SDS se une en una proporción de aproximadamente 1,4 g SDS por 1,0 g de proteína (aunque las proporciones de unión pueden variar entre 1,1 y 2,2), proporcionando una relación carga/masa aproximadamente uniforme para la mayor parte de las proteínas, de modo que se puede asumir que la distancia de migración en el gel está directamente vinculada sólo al tamaño de la proteína desplegada (longitud de su cadena, número de aminoácidos o masa molecular). Se puede añadir un colorante trazador a la disolución de proteínas para permitir al experimentador seguir el curso de la electroforesis.
Ingredientes químicos y sus papeles
El gel de poliacrilamida era ya conocido como un medio potencial para la inclusión de tejidos seccionados en 1959.[4] [5] Se trata de un gel sintético, termoestable, transparente, resistente, relativamente inerte, químicamente hablando, que puede prepararse con un amplio intervalo de tamaños medios de poro.[6] El tamaño de poro de un gel está determinado por dos factores: la concentración total de acrilamida presente (%T) (T = concentración total de monómeros de acrilamida y bisacrilamida) y la cantidad del reticulador (%C) (C = concentración del reticulador). Los tamaños de poro disminuyen cuando aumenta %T, y con reticulación el 5%C produce el menor tamaño de poro. Cualquier incremento o disminución en %C aumenta el tamaño de poro (función parabólica). Esto parece deberse a la estructura no homogénea de los filamentos del gel.
El gel formado a partir de este material también puede resistir gradientes de alto voltaje, ser viable para varios procedimientos de tinción y destinción y puede ser digerido para extraer fracciones separadas o desecado para su autorradiografía y registro permanente. La electroforesis en disco utiliza geles de distintos tamaños de poro.[7] [8] El nombre de electroforesis en disco deriva del acrónimo inglés DISC, que hace referencia a las discontinuidades en la matriz electroforética y por coincidencia a la forma discoidal de las zonas separadas de iones. Hay dos capas de gel, el gel de apilamiento o espaciador y el gel de resolución o separador.
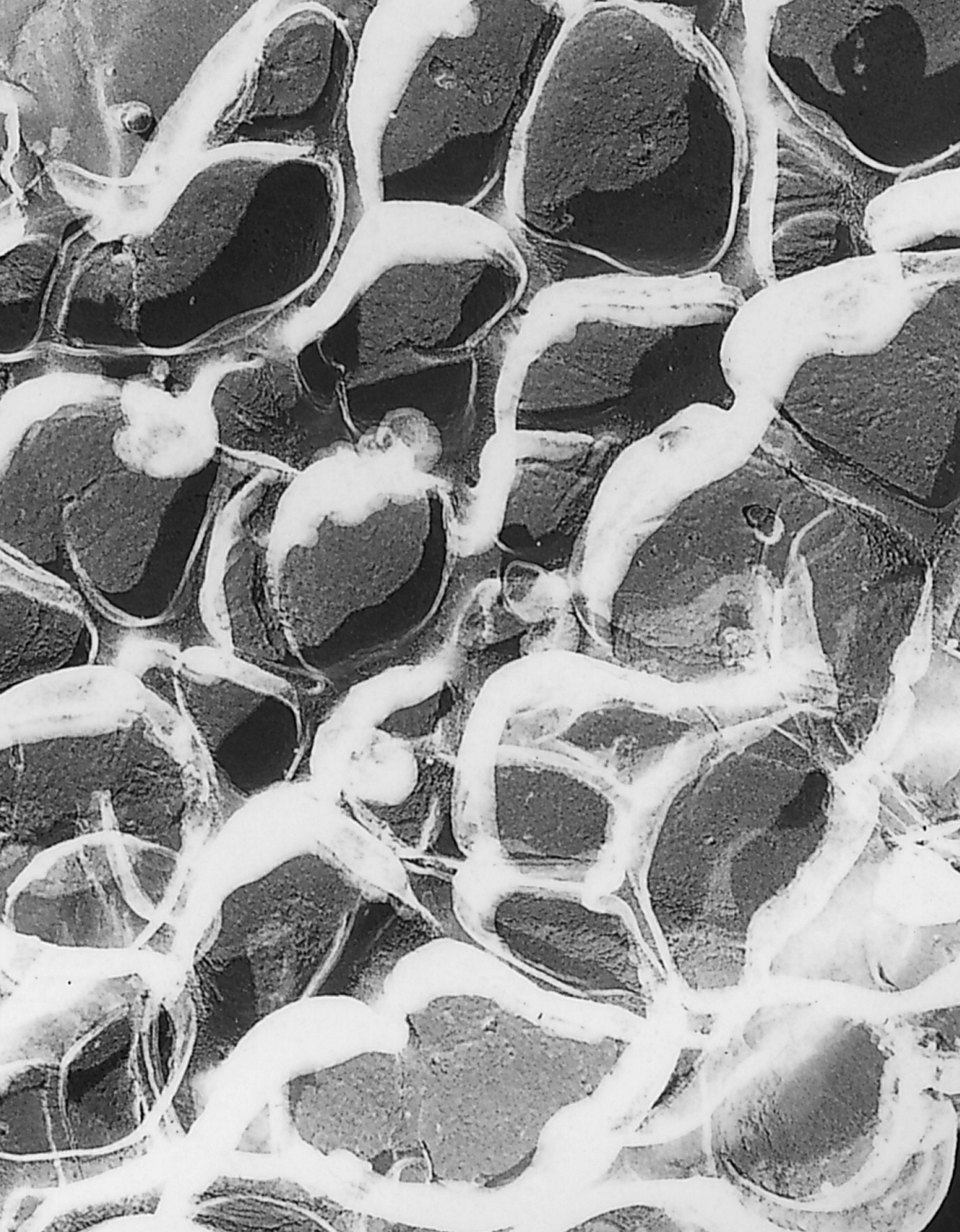
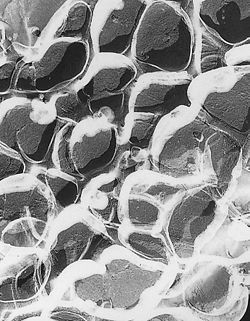 Imagen de Microscopía electrónica de transmisión de un gel de poliacrilamida. El tamaño de poro de un gel esta determinado por la cantidad total de monómero presente (%T) y la cantidad de reticulador (%C).
Imagen de Microscopía electrónica de transmisión de un gel de poliacrilamida. El tamaño de poro de un gel esta determinado por la cantidad total de monómero presente (%T) y la cantidad de reticulador (%C).Gel de apilamiento
El gel de apilamiento es un gel de poliacrilamida con tamaño de poro grande (4%T). Este gel está preparado con un tampón de Tris/HCl con un pH de 6,8, dos unidades de pH menor que el tampón de electroforesis (Tris/glicina). Estas condiciones proporcionan un ambiente para las reacciones de Kohlrausch que determinan la conductividad molar, y como resultado, las proteínas recubiertas con SDS se concentran varias veces, resultando así en pocos minutos una zona de inicio del orden de 19 μm. Este gel se coloca sobre el gel de resolución. La altura de la zona correspondiente al gel de apilamiento debe ser superior al doble de la altura y el volumen de la muestra que va a ser aplicada.
Gel de resolución
El gel de resolución es un gel de poliacrilamida de poro pequeño (3 - 30% de monómero de acrilamida) que típicamente se confecciona utilizando un tampón Tris/HCl con un pH de 8,8. En el gel de resolución es donde las macromoléculas se separan de acuerdo a su tamaño. Los geles de resolución tienen un intervalo óptimo de separación dependiente del porcentaje de monómero utilizado para su preparación. Por ejemplo, se pueden utilizar eficazmente geles con contenidos de 8%, 10% y 12% para separar proteínas de 24 a 205 kDa, 14 a 205 kDa, y 14 a 66 kDa, respectivamente.
Ingredientes químicos
- Tris, nombre abreviado del tris (hidroximetil) aminometano (C4H11NO3; Mr = 121,14). Se utiliza a menudo como tampón porque es una sustancia inocua para la mayor parte de las proteínas. Su pKa es 8,3 a 20 °C, lo que lo convierte en un tampón idóneo para el intervalo de pH entre 7 y 9.
- Glicina, o ácido aminoacético (C2H5NO2; Mr = 75,07). Se ha utilizado como fuente de iones de arrastre (o lentos) porque su pKa es 9,69 y la movilidad del glicinato es tal que se puede situar en un valor inferior a la de la proteína más lenta conocida con carga negativa neta para el intervalo de pH de trabajo. El mínimo de este intervalo está situado aproximadamente en 8,0.
- Acrilamida (C3H5NO; Mr = 71,08). Es un sólido pulverulento, blanco y cristalino. Cuando se encuentra en disolución acuosa experimenta autopolimerización de forma espontánea y lenta; el resultado es que las moléculas de acrilamida se unen cabeza con cola. En presencia de un sistema generador de radicales libres, los monómeros de acrilamida se activan, quedando ellos mismos en estado de radical libre, y reaccionan rápidamente para formar polímeros de cadena larga. Este tipo de reacción se conoce como polimerización vinílica. Sin embargo, el polímero en disolución no forma un gel, sino que se encuentra en estado viscoso, debido a que las cadenas pueden deslizar unas sobre otras. La formación del gel requiere que varias cadenas queden trabadas (lo que se consigue con la bisacrilamida). La acrilamida es una neurotoxina, por lo que debe ser manejada con precaución. También es esencial almacenarla en un lugar refrigerado, seco y oscuro para reducir la autopolimerización y la hidrólisis.
- Bisacrilamida, o N,N'-metilenbisacrilamida (C7H10N2O2; Mr = 154,17). La bisacrilamida es el agente de reticulación más frecuente para los geles de poliacrilamida. Su estructura está compuesta por dos moléculas de poliacrilamida enlazadas por sus grupos amino, no reactivos de cara a la polimerización. Por ello, la bisacrilamida polimeriza conjuntamente con la acrilamida pero establece puentes entre las cadenas lineales de poliacrilamida, y así evita el deslizamiento de éstas y conduce a la formación del gel.
- Dodecilsulfato sódico (SDS) (C12H25NaO4S; Mr = 288,38). Se trata del agente disociador más habitual para desnaturalizar proteínas nativas en sus polipéptidos individuales. Cuando se calienta brevemente una mezcla de proteínas a 100ºC en presencia de SDS, el detergente recubre el polipéptido alrededor de su eje central, manteniéndolo desplegado. En este proceso, las cargas intrínsecas del polipéptido son despreciables en comparación por las aportadas por el SDS. De este modo los polipéptidos se transforman después del tratamiento en estructuras con forma de bastón que poseen una densidad de carga uniforme en toda su longitud. La movilidad de estas proteínas es una función aproximadamente lineal del logaritmo de su masa molecular.
- Persulfato amónico (APS) (N2H8S2O8; Mr = 228,2). El APS genera radicales libres y es por ello un iniciador de la reacción de polimerización que finalmente forma el gel.
- TEMED, acrónimo de N,N,N',N'-tetrametiletilenodiamina (C6H16N2; Mr = 116,21). Se trata de otro iniciador (propagador). La tasa de polimerización y las propiedades del gel resultante dependen de la concentración de APS y TEMED. Aumentando su contenido se disminuye la longitud media de la cadena de polímero y se incrementa la turbidez del gel, al tiempo que disminuye su elasticidad. Por contra, disminuyendo la cantidad de iniciadores se obtiene el efecto inverso. Se debe, por tanto, utilizar la menor concentración posible de catalizadores que permita la polimerización en un tiempo óptimo. Se utiliza APS y TEMED en concentraciones equimolares del orden de 1 a 10 mM.
Reactivos para el procesamiento y la visualización
Se utilizan los siguientes reactivos para el procesamiento del gel y las muestras de proteínas que se visualizan en el:
- Azul de bromofenol (BPB, de bromophenol blue) (3',3",5',5"-tetrabromofenolsulfoftaleína) (C19H10Br4O5S; Mr = 669,99). Se trata del colorante utilizado más frecuentemente como marcador de avance en electroforesis. Las proteínas y los ácidos nucleicos son mayormente incoloros. Cuando están sometidos a electroforesis es importante detener el avance antes de que sobrepasen el extremo del gel. El azul de bromofenol es el colorante trazador más empleado porque es viable en pH neutro y alcalino, es una molécula pequeña, es ionizable y posee carga negativa a pH superior a 4,6, de modo que migra al ánodo. Al ser una molécula pequeña se adelanta a la mayor parte de las proteínas y ácidos nucleicos. Cuando el experimentador observa que el colorante alcanza el extremo anódico del gel, debe desconectar la corriente y dar por terminada la separación. Se puede unir débilmente a las proteínas, dotándolas de color azul.
- Glicerol (C3H8O3; Mr = 92,09). Debido a su densidad, contribuye a que la muestra quede depositada en los pocillos sin dispersarse.
- Coomassie Blue (C45H44N3NaO7S2; Mr = 825,97). El CBB (de Coomassie Brilliant Blue, azul brillante de Coomassie) es el colorante para proteínas más popular. Es de tipo aniónico, y se une a proteínas de modo inespecífico. Su estructura es predominantemente no polar, por lo que habitualmente se usa (al 0,025%) en disolución con metanol (40%) y acético (7%). Las proteínas del gel se fijan gracias al ácido acético y al mismo tiempo se tiñen. El colorante en exceso que se incorpora en el gel se puede eliminar destiñendo con una disolución de composición idéntica excepto por el colorante. Las proteínas se detectan como bandas azules contra un fondo claro. Puesto que el SDS también es aniónico, puede interferir con el proceso de tinción. Por tanto, se recomienda un gran volumen de disolución de tinción, aproximadamente diez veces el volumen del gel.
- Butanol (C4H10O; Mr = 74,12). Se utiliza butanol saturado con agua como disolución de recubrimiento en el gel de resolución.
- 2-Mercaptoetanol (HS-CH2CH2OH; Mr = 78,13). El 2-mercaptoetanol es un agente reductor que se utiliza para romper los enlaces disulfuro y asegurarse de que la proteína está completamente desnaturalizada antes de ser cargada en el gel, y de ese modo garantizar que corre de manera uniforme.
SDS-PAGE reductora
Además de la adición de SDS, se puede calentar opcionalmente las proteínas durante un breve tiempo a una temperatura próxima a la de ebullición en presencia de un agente reductor, como el ditiotreitol (DTT) o el 2-mercaptoetanol (o beta-mercaptoetanol), que contribuirá a desnaturalizar las proteínas reduciendo los enlaces disulfuro, desplegando así algunas formas de plegamiento terciario y rompiendo la estructura cuaternaria (subunidades oligoméricas). Esto es lo que se conoce como SDS-PAGE reductora, y se utiliza de manera habitual. Este procedimiento no se usa cuando la estructura nativa es importante para análisis posteriores (p.ej. actividad enzimática, que se muestra mediante el uso de zimogramas). Un ejemplo sería el QPNC-PAGE, (de Quantitative Preparative Native Continuous PAGE, en inglés: PAGE de preparación, cuantitativa, nativa y continua). Es un nuevo método para la separación de metaloproteínas nativas en matrices biológicas.
Electroforesis y tinción
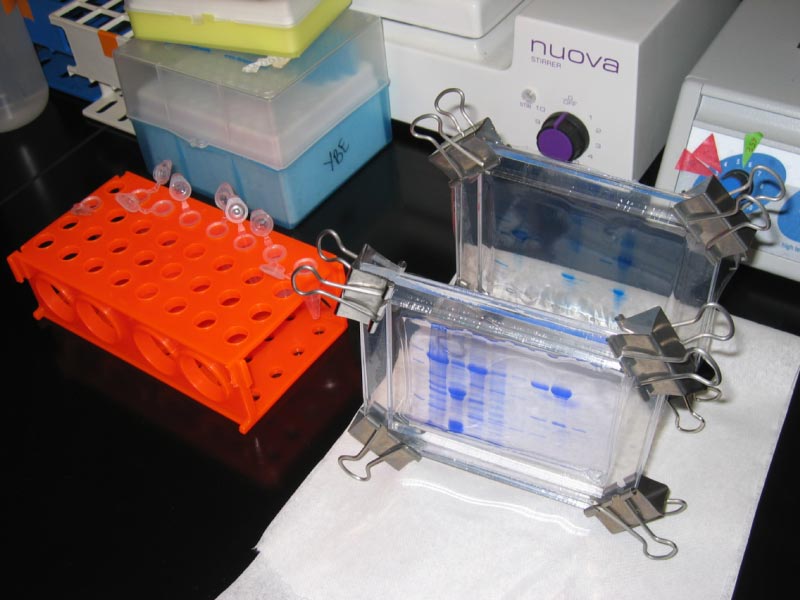
 Dos geles SDS-PAGE tras completar la electroforesis.
Dos geles SDS-PAGE tras completar la electroforesis.Las proteínas desnaturalizadas son posteriormente aplicadas en un extremo de una capa del gel de poliacrilamida en un tampón químico adecuado. Se aplica una corriente eléctrica que recorre el gel, provocando que las proteínas con carga negativa migren a través de él en dirección al ánodo. Dependiendo de su tamaño, cada proteína se moverá de modo diferente a través de la matriz del gel: las proteínas de tamaño corto encajarán más fácilmente a través de los poros del gel, mientras que las de tamaño mayor tendrán mayor dificultad para hacerlo (encontrarán mayor resistencia). Tras un cierto tiempo (habitualmente unas horas, aunque esto depende del voltaje aplicado al gel: se debe tener que voltajes mayores producen una migración más rápida, pero tienden a producir una resolución algo más deficiente), las proteínas habrán migrado de forma diferencial en base a su tamaño. Las menores se desplazarán más rápido, mientras que las mayores permanecerán más cerca del punto de origen. Por tanto, las proteínas se separarán aproximadamente de acuerdo a su tamaño (y por tanto, su peso moleculasr). Tras la electroforesis, el gel debe ser teñido (lo más habitual con Coomassie blue o tinción argéntea, permitiendo la visualización de las proteínas separadas o su procesamiento posterior (ej.: Western blot). Tras la tinción, las diferentes proteínas apareceran como bandas distintas en el gel. Es común correr marcadores moleculares de tamaño molecular conocido en una calle a parte del gel para calibrarlo y determinar el peso de proteínas desconocidas comparando la distancia recorrida con la del marcador. El gel se forma en realidad porque la solución contiene una pequeña cantidad, normalmente 1 parte en 35 de bisacrilamida, que puede formar enlaces cruzados entre dos moléculas de poliacrilamida. La proporción entre ambas sustancias puede variar de acuerdo al propósito para el que se diseña.
La electroforesis en gel es normalmente la primera opción en un ensayo de pureza de proteínas debido a su fiabilidad y a su sencillez. Aún así se pueden dar falsos positivos y negativos. Un contaminante que migre conjuntamente puede aparecer en la misma banda que la proteína deseada. Esta comigración también puede hacer que la proteína migre en una posición diferente o no sea capaz de penetrar en el gel. Esto es por lo que es importante teñir el gel completo, incluida la sección de apilamiento (o carga). El colorante Coomassie Blue también puede unirse con menor afinidad a las glicoproteínas y proteínas fibrosas, lo que interfiere con la cuantificación.
Tinción con plata
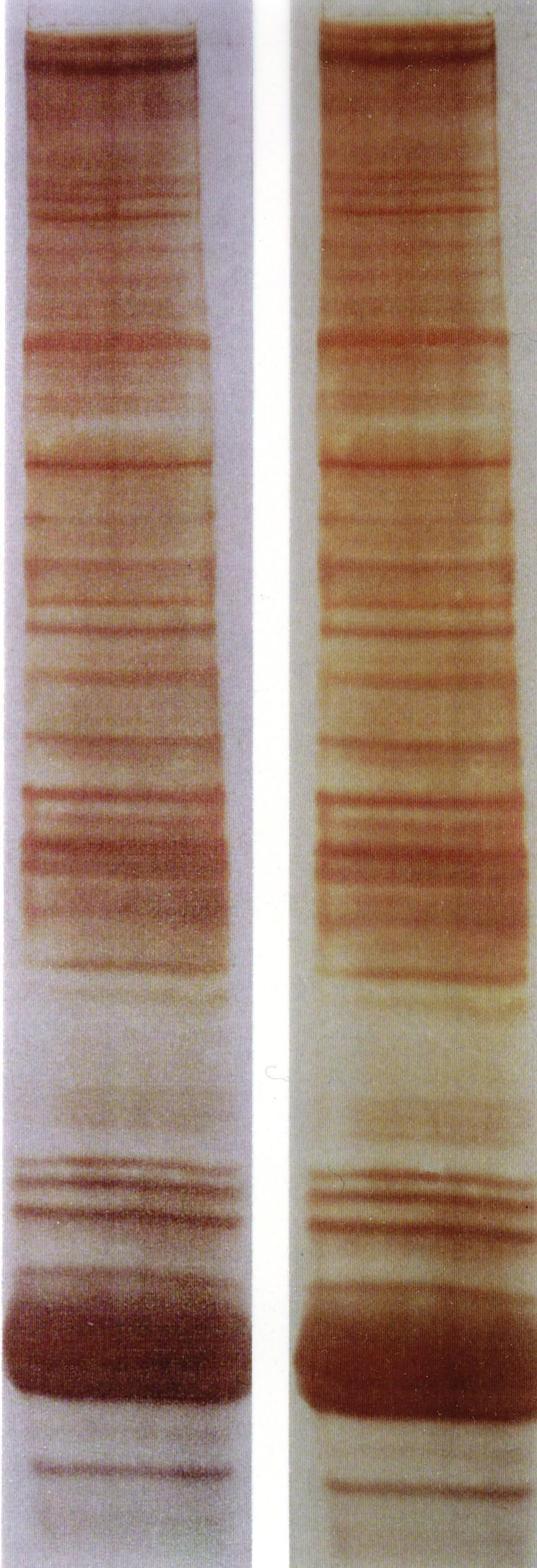
 SDS- PAGE con tinción con plata.
SDS- PAGE con tinción con plata.
En el siglo XIV la técnica de la tinción con plata se desarrolló para colorear superficies de cristal. Fue ampliamente utilizada para este propósito hasta el siglo XVI. El color producido por las primeras tinciones variaba entre un amarillo claro y un naranja rojizo. Camillo Golgi perfeccionó la tinción con plata para el estudio del sistema nervioso. El método de Golgi tiñe por completo un número limitado de células al azar. El mecanismo químico exacto por lo que esto sucede aún es ampliamente desconocido.[9] Kerenyi y Gallyas introdujeron esta tinción como un procedimiento sensible para detectar cantidades traza de proteínas en geles.[10] La técnica se extendió al estudio de otras macromoléculas biológicas tras su separación por varios soportes.[11] La tinción de Coomassie Blue clásica puede detectar normalmente bandas de proteína de 50 ng, mientras que la tinción con plata incrementa este límite de sensibilidad en unas 50 veces. Muchas variables pueden influir en la intensidad del color y todas las proteínas tienen características distintas de tinción. Para una buena tinción se debe prestar atención a la limpieza del material de vidrio, la pureza del agua y de los reactivos.[12]Sistemas de tamponamiento químico
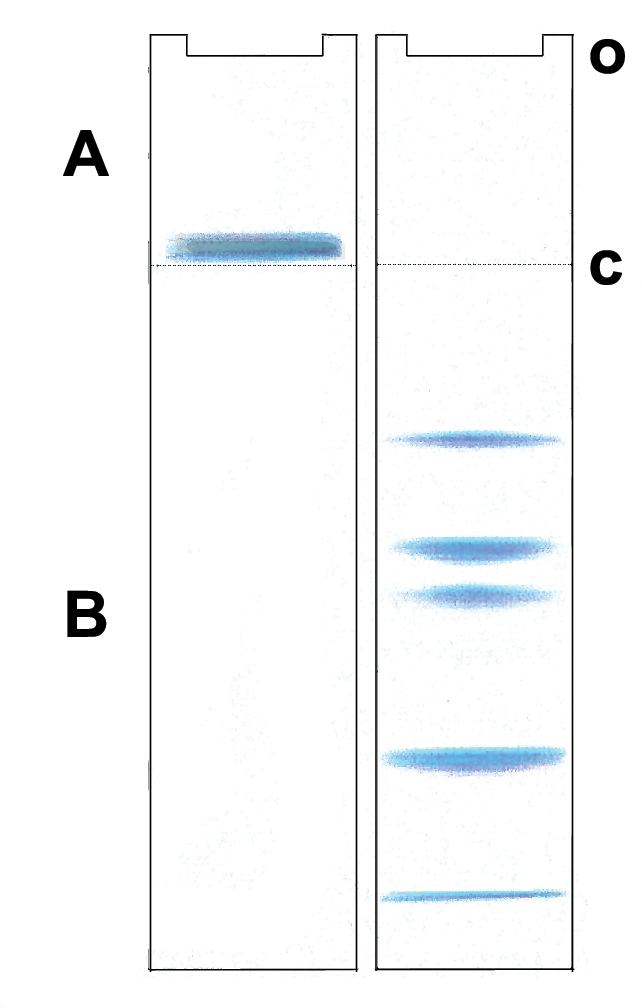
 Migración de proteínas postulada en el sistema de gel Laemmli. A: Gel de carga, B: Gel de resolución o: aplicación de la muestra c: discontinuidades en la matriz electroforética y del tampón químico.
Migración de proteínas postulada en el sistema de gel Laemmli. A: Gel de carga, B: Gel de resolución o: aplicación de la muestra c: discontinuidades en la matriz electroforética y del tampón químico.La mayoría de las separaciones de proteínas se llevan a cabo utilizando un sistema de tampón "discontinuo" en disolución que incrementa significativamente la delgadez de las bandas en el gel. Durante la electroforesis en un sistema de gel discontinuo, se forma un gradiente iónico en los estadios tempranos de la electroforesis que hacen que todas las proteínas se enfoquen en una sola banda estrecha. Esto sucede en una región del gel que tiene poros más grandes de modo que la matriz del gel no retarda la migración en el momento del enfocado o "apilamiento". Los iones negativos del tampón contenido en el tanque "sobrepasa" el "paquete" de proteínas recubiertas con SDS y eliminan el gradiente iónico de modo que posteriormente las proteínas se separan por la acción de tamiz en la región del gel que está más abajo, la de "resolución".
Muchas personas continúan utilizando el sistema de tamponamiento de tris-glicina, también llamado de "Laemmli" que apila en un pH de 6,8 y resuelve a Many people continue to use a tris-glycine or "Laemmli" buffering system that stacks at a ~8.3-9.0. Estos niveles de pH favorecen la formación de enlaces disulfuro entre los residuos de cisteína en las proteínas, especialmente cuando se encuentran presentes a elevadas concentraciones, debido a que los rangos de pKa de cisteína varían entre 8-9 y también debido a que el agente reductor presente en el buffer de carga no migra conjuntamente con las proteínas. Los avances recientes en la tecnología de tamponamiento minorizan este problema resolviendo las proteínas a un pH muy por debajo del pKa de cisteína (p.ej., bis-tris, pH 6.5) y contienen también agentes reductores (p.ej. bisulfito de sodio) que se mueven en el gel por delante de las proteínas para mantener un ambiente reductor. Un beneficio adicional de utilizar tampones con pH más bajo es la mayor estabilidad del gel de acrilamida, de modo que los geles se pueden almacener durante periodos de tiempo mayores antes de su uso.[13] [14]
Electroforesis de proteínas en gradiente por SDS
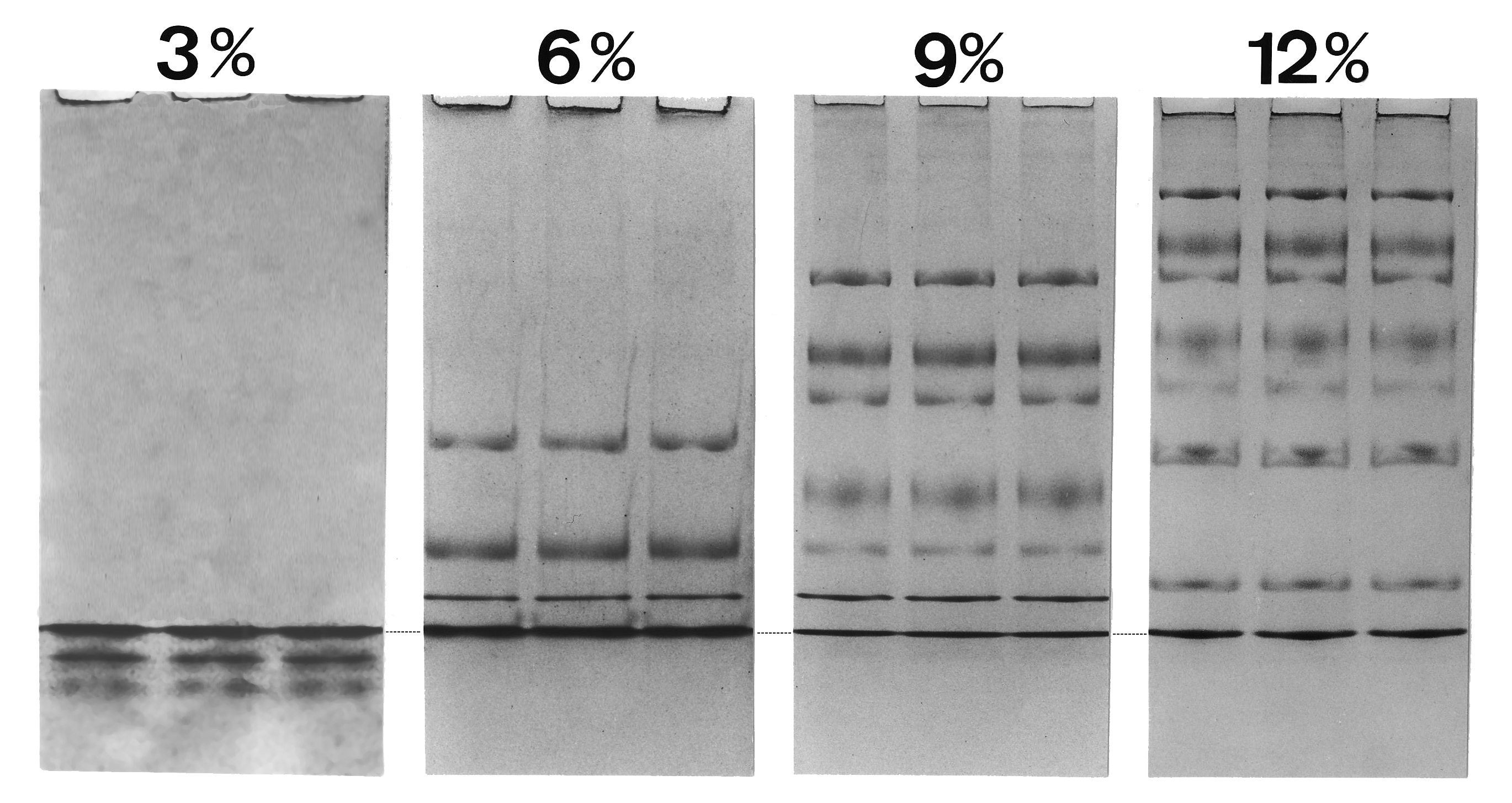
 Migración de proteínas en geles de SDS a distintas concentraciones de acrilamida (%T). Se muestra la migración de nueve proteínas que van desde 94 kDa a 14.4 kDa. El apilamiento y descompactación se suceden de modo contínuo en el gel, en cada proteína con distinta concentración del gel. La línea de puntos indica la discontinuidad en la interfase móvil Gly¯/Cl¯. Las Proteínas entre el electrolito director rápido y el electrolito lento de arrastre no se diluyen por difusión.
Migración de proteínas en geles de SDS a distintas concentraciones de acrilamida (%T). Se muestra la migración de nueve proteínas que van desde 94 kDa a 14.4 kDa. El apilamiento y descompactación se suceden de modo contínuo en el gel, en cada proteína con distinta concentración del gel. La línea de puntos indica la discontinuidad en la interfase móvil Gly¯/Cl¯. Las Proteínas entre el electrolito director rápido y el electrolito lento de arrastre no se diluyen por difusión.A medida que se aplica un voltaje, los aniones (y las muestras de moléculas cargadas negativamente) migran hacia el electrodo positivo en la cámara inferior, y el ión conductor es el Cl¯ (alta movilidad y elevada concentración); el glicinato es el ión de arrastre (baja movilidad y elevada concentración). Las partículas de SDS-proteína no migran libremente entre el borde de Cl¯ odel tampón del gel y las de Gly¯ del tampón del cátodo. Friedrich Kohlrausch encontró que la ley de Ohm también se aplica a los electrolitos en disolución. Puesto que el voltaje cae entre los buffers de cloro y glicina, las proteínas se comprimen (apilan) en finas capas de micrómetros.[15] El frente se mueve a través de un gradiente de poro y el paquete de proteínas se dispersa gradualmente debido a una resistencia por fricción cada vez mayor en la matriz del gel. Se da contínuamente una compresión y descompresión en el gradiente del gel en distinta posición para cada proteína. Para que se produzca un desempaquetamiento completo la concentración de poliacrilamida debe superar el 16% T. El sistema de dos geles de "Laemmli" es simplemente un gel en gradiente de poro. La discontinuidad de pH de los tampones no tiene importancia para la calidad de la separación, y no se precisa un gel de apilamiento con diferente pH.
Véase también
- Electroforesis capilar
- Electroforesis en gel
- Electroforesis proteica
- Electroforesis
- Isoelectroenfoque
- Northern blot
- Southern blot
- Western blot
Referencias
- ↑ Shapiro AL, Viñuela E, Maizel JV Jr. (September de 1967). «Molecular weight estimation of polypeptide chains by electrophoresis in SDS-polyacrylamide gels.» Biochem Biophys Res Commun.. Vol. 28. n.º 5. pp. 815-820. PMID 4861258.
- ↑ Weber K, Osborn M (August de 1969). «The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis.» J Biol Chem.. Vol. 244. n.º 16. pp. 4406-4412. PMID 5806584.
- ↑ Laemmli UK (August de 1970). «Cleavage of structural proteins during the assembly of the head of bacteriophage T4» Nature. Vol. 227. n.º 5259. pp. 680–685. PMID 5432063.
- ↑ Davis BJ, Ornstein L (1959). «A new high resolution electrophoresis method.» Delivered at the Society for the Study of Blood at the New York Academy of Medicine.
- ↑ Raymond S, Weintraub L. (1959). «Acrylamide gel as a supporting medium for zone electrophoresis.» Science. Vol. 130. pp. 711. PMID 14436634.
- ↑ Rüchel R, Steere RL, Erbe EF (1978). «Transmission-electron microscopic observations of freeze-etched polyacrylamide gels.» J Chromatogr.. Vol. 166. pp. 563-575.
- ↑ Ornstein L (December de 1964). «DISC ELECTROPHORESIS. I. BACKGROUND AND THEORY.» Ann N Y Acad Sci.. Vol. 121. pp. 321-349. PMID 14240533.
- ↑ Davis BJ (December de 1964). «Disc Electrophoresis. 2, Method and application to human serum proteins» Ann. New York Acad. Sci. Vol. 121. pp. 404-427. PMID 14240539.
- ↑ Golgi C (1873). «Sulla struttura della sostanza grigia del cervello.» Gazzetta Medica Italiana (Lombardia). Vol. 33. pp. 244–246.
- ↑ Kerenyi L, Gallyas F (1973). «Über Probleme der quantitiven Auswertung der mit physikalischer Entwicklung versilberten Agarelektrophoretogramme» Clin. Chim. Acta. Vol. 47. pp. 425-436.
- ↑ Switzer RC 3rd, Merril CR, Shifrin S (Sep de 1979). «A highly sensitive silver stain for detecting proteins and peptides in polyacrylamide gels.» Anal Biochem.. Vol. 98. n.º 1. pp. 231-237. PMID 94518.
- ↑ Hempelmann E, Schulze M, Götze O (1984). «Free SH-groups are important for the polychromatic staining of proteins with silver nitrat» Neuhof V (ed)Electrophoresis '84 , Verlag Chemie Weinheim 1984. pp. 328-330.
- ↑ Schägger H, von Jagow G (1987). «Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa.» Anal Biochem.. Vol. 166. n.º 2. pp. 368-379. PMID 2449095.
- ↑ Wiltfang J, Arold N, Neuhoff V (1991). «A new multiphasic buffer system for sodium dodecyl sulfate-polyacrylamide gel electrophoresis of proteins and peptides with molecular masses 100,000-1000, and their detection with picomolar sensitivity.» Electrophoresis. Vol. 12. n.º 5. pp. 352-366. PMID 1718736.
- ↑ Kohlrausch F (1897). «Ueber Concentrations-Verschiebungen durch Electrolyse im Inneren von Lösungen und Lösungsgemischen.» Ann.J.Phys.u.Chem.. Vol. 62. pp. 209-239.
Enlaces externos
- Video sobre SDS-PAGE
- Calculador de SDS-PAGE para protocolos de geles Tris-Urea.
Categorías: Biología molecular | Técnicas de laboratorio
Wikimedia foundation. 2010.