- Síndrome de Bernard-Soulier
-
Síndrome de Bernard-Soulier Clasificación y recursos externos CIE-10 D69.1 CIE-9 287.1 OMIM 231200 DiseasesDB 1356 eMedicine ped/230 MeSH D001606  Aviso médico
Aviso médicoEl Síndrome de Bernard-Soulier también llamado distrofia trombocítica hemorrágica[1] es una rara enfermedad genética de herencia autosómica recesiva que afecta la correcta coagulación debido a la deficiencia de la glicoproteína Ib, receptor para el factor de von Willebrand, alterando de esta forma la hemostasia primaria.
Se estima que la incidencia es menor a 1 en un millón de personas, basado en casos de Europa, Norteamérica y Japón.[2] Se caracteriza además por tener plaquetas de gran tamaño (megacariocitos)
Contenido
Etiología
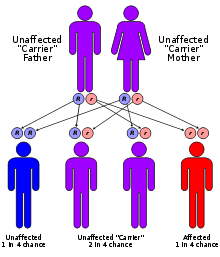 El síndrome tiene una herencia de tipo autosómica recesiva
El síndrome tiene una herencia de tipo autosómica recesiva
El origen del síndrome es de causa genética, existiendo alteración en las proteína de la membrana plaquetelar. Existen tres formas[3]
- Tipo A - GP1BA
- Tipo B - GP1BB
- Tipo C - GP9
Presentación
Se caracteriza por un tiempo de sangría prolongado, trombocitopenia, macroplaquetas y disminución en la vida media plaquetelar. El síndrome está asociado con déficit cuantitativos y cualitativos del complejo glicoprotéico GP1b/V/IX. El defecto produce la incapacidad de que las plaquetas se agreguen y se unan a los sitios de la lesión endotelial.[4] El grado de trombocitopenia puede ser estimado incorrectamente debido a que el conteo de plaquetas se hace por procesos automatizados en donde al existir plaquetas de gran tamaño pueden no ser cuantificadas. Típicamente, las plaquetas del síndrome de Bernard-Soulier no se agregan con ristocetina y este defecto no es corregido con la adición de plasma, con lo cual se diferencia de la enfermedad de Von Willebrand.
Síntomas
- Epistaxis
- Gingivorragia
- Ser propenso a equimosis
- Metrorragia
- Sangrado prolongado posterior a intervención quirúrgica
Diagnóstico
El diagnóstico se basa en la clínica más las pruebas de:
- Tiempo de Sangrado (orienta el diagnóstico)
- Frotis con macroplaquetas
- Prueba de agregación con ristocetina que no responde a la adición de plasma.
Tratamiento
El evento es sintomático. Los eventos de sangrado pueden ser muy severos, teniendo que controlarse con transfusiones de plaquetas. La mayoría de los heterocigotos, con muy pocas excepciones, no sufren de sangrado fácil.
Epónimo
El síndrome lleva el nombre del Dr Jean Bernard y Jean Pierre Soulier[5] [6]
Referencias
- ↑ Lanza F (2006). «Bernard-Soulier syndrome (hemorrhagiparous thrombocytic dystrophy)». Orphanet J Rare Dis. 16 (1): pp. 46. doi:. PMID 17109744.
- ↑ Anesthetic and perioperative management of a patient with Bernard-Soulier syndrome Georgia Kostopanagiotou MD, Ioanna Siafaka MDa, Constantinos Sikiotis MDa, and Vassilios Smyrniotis MDa. Received 23 July 2003; Revised 21 October 2003
- ↑ OMIM 231200
- ↑ Pham A, Wang J (2007). «Bernard-Soulier syndrome: an inherited platelet disorder» (subscription required). Arch. Pathol. Lab. Med. 131 (12): pp. 1834–6. PMID 18081445. http://journals.allenpress.com/jrnlserv/?request=get-abstract&issn=0003-9985&volumen=131&page=1834.
- ↑ synd/2075 en Who Named It?
- ↑ Bernard J, Soulier JP (December 1948). «[Sur une nouvelle variété de dystrophie thrombocytaire hémorragipare congénitale]» (en French). Semaine des hôpitaux de Paris 24 (Spec. No.): pp. 3217–23. PMID 18116504.
Categorías:- Enfermedades raras
- Hematología

Wikimedia foundation. 2010.