- Coagulación
-
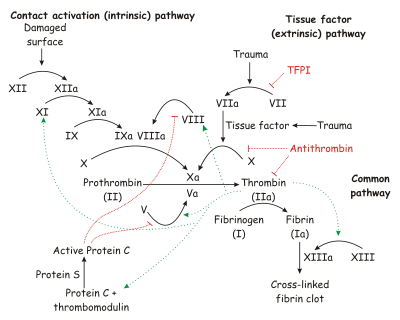 La cascada completa de coagulación. En el texto se describen las diferentes vías y factores de coagulación.
La cascada completa de coagulación. En el texto se describen las diferentes vías y factores de coagulación.
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre.
Se denomina coagulación al proceso, por el cual, la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoléculares en forma de una red tridimensional.
El fibrinógeno, una vez transformado, recibe el nombre de fibrina. Coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse.
Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas.
Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
Contenido
Factores de coagulación
El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc.
En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares.
A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (precalicreína —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada.
Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para su síntesis en el hígado, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
Factor Nombre Masa (KDa) Nivel en plasma (mg/dl) Función I Fibrinógeno 340 250-400 Se convierte en fibrina por acción de la trombina. La fibrina constituye la red que forma el coágulo. II Protrombina 72 10-14 Se convierte en trombina por la acción del factor Xa. La trombina cataliza la formación de fibrina a partir de fibrinógeno. III Factor tisular de tromboplastina Se libera con el daño celular; participa junto con el factor VIIa en la activación del factor X por la vía extrínseca. IV Ion Calcio 40 Da 4-5 Median la unión de los factores IX, X, VII y II a fosfolípidos de membrana. V proacelerina (leiden) 350 1 Potencia la acción de Xa sobre la protrombina VI Variante activada del factor V -- -- -- VII Proconvertina 45-54 0.05 Participa en la vía extrínseca, forma un complejo con los factores III y Ca2+ que activa al factor X. VIII:C Factor antihemofílico 285 0.1-0.2 Indispensable para la acción del factor X (junto con el IXa). Su ausencia provoca hemofilia A. VIII:R Factor Von Willebrand >10000 Media la unión del factor VIII:C a plaquetas. Su ausencia causa la Enfermedad de Von Willebrand. IX Factor Christmas 57 0.3 Convertido en IXa por el XIa. El complejo IXa-VII-Ca2+ activa al factor X. Su ausencia es la causa de la hemofilia B. X Factor Stuart-Prower 59 1 Activado por el complejo IXa-VIII-Ca2+ en la vía intrinseca o por VII-III-Ca2+ en la extrínseca, es responsable de la hidrólisis de protrombina para formar trombina. XI Tromboplastina plasmática o antecedente trombo plastínico de plasma 160 0.5 Convertido en la proteasa XIa por acción del factor XIIa; XIa activa al factor IX. Su ausencia es la causa de la hemofilia C. XII Factor Hageman 76 -- Se activa en contacto con superficies extrañas por medio de calicreína asociada a quininógeno de alto peso molecular; convierte al factor XI en XIa. XIII Pretransglutaminidasa o factor Laili-Lorand 320 1-2 Activado a XIIIa, también llamado transglutaminidasa, por la acción de la trombina. Forma enlaces cruzados entre restos de lisina y glutamina contiguos de los filamentos de fibrina, estabilizándolos. Precalicreína Factor Fletcher -- -- Activada a calicreína, juntamente con el quininógeno de alto peso molecular convierte al factor XII en XIIa. quininógeno de alto peso molecular Factor Fitzgerald-Flaujeac-Williams -- -- Coayuda con la calicreína en la activación del factor XII. Etapas de la cascada de coagulación
 Resumen de la cascada de coagulación
Resumen de la cascada de coagulaciónLa cascada de coagulación se divide para su estudio, clásicamente en tres vias: La via intrínseca, la vía extrínseca y la vía común.
Las vías intrínseca y extrínseca son las vías de iniciación de la cascada, mientras que la vía común es hacia donde confluyen las otras dos desembocando en la conversión de fibrinógeno en fibrina.
Esta división es un tanto arbitraria y tiene más que ver con las deficiencias de las técnicas que en su momento se utilizaron para desentrañar los mecanismos implicados, que con lo que ocurre realmente en una lesión vascular; ya que en este último caso se establecen varias interrelacciones entre las vías de iniciación.
Mecanismo básico
Cada reacción de estas vías da como resultado el ensamblado de un complejo compuesto por una enzima (factor de coagulación activado), un sustrato (proenzima de un factor de coagulación) y un cofactor que actúa posibilitando la reacción.
Estos componentes se ensamblan en general sobre una superficie fosfolipídica y se mantienen unidos por medio de puentes formados por iones Ca2+. Por lo tanto la reacción en cascada tiende a producirse en un sitio donde este ensamblaje puede ocurrir; por ejemplo sobre la superficie de plaquetas activadas.
Tanto la vía intrínseca como la vía extrínseca desembocan en la conversión del factor X en Xa (la letra "a" como subíndice "a" significa "activado") punto en el que se inicia la vía común.
Vía intrínseca
Recibe este nombre debido a que antiguamente se pensaba que la sangre era capaz de coagular "intrínsecamente" por esta vía sin necesidad de contar con la ayuda de factores externos. Actualmente se sabe que esto no es exactamente así. De hecho la vía extrínseca es la que realmente inicia el proceso y la vía intrínseca sirve de amplificación y seguridad del proceso homeostático.
El proceso de coagulación en esta vía se desencadena cuando la sangre entra en contacto con una superficie "extraña", es decir, diferente al endotelio vascular.
En el caso de una lesión vascular, la membrana basal del endotelio o las fibras colágenas del tejido conectivo, proporcionan el punto de iniciación.
En general las superficies polianiónicas (cargadas negativamente) pueden cumplir el mismo papel, tanto materiales orgánicos como la celulosa, o no orgánicos como el vidrio, el caolín o algunas resinas pueden actuar como desencadenantes de la reacción.
A esta vía es posible subdividirla en tres pasos:
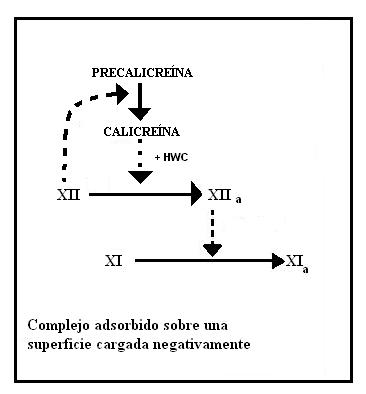
Formación del factor XIa
En esta etapa participan cuatro proteínas: Precalicreína, Quininógeno de alto peso molecular (HMWK) y los factores XII y XI. Esta etapa no requiere de iones calcio.
Estos cuatro factores se adsorben sobre la superficie cargada negativamente, formando el complejo cebador o de iniciación. De estos factores el XII funciona como verdadero iniciador, ya que si bien es una proenzima, posee una pequeña actividad catalítica que alcanza para activar a la precalicreína convirtiéndola en calicreína.
En segunda instancia la calicreína actúa catalíticamente sobre el factor XII para convertirlo en XIIa, una enzima muchísimo más activa. La actividad catalítica de la calicreína se ve potenciada por el HMWK.
Por último la proteasa XIIa actúa sobre el factor XI para liberar XIa.
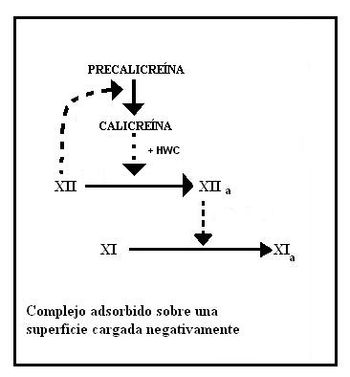 Activación del factor XI
Activación del factor XIFormación del factor IXa
El factor IX se encuentra en el plasma como una proenzima. En presencia de iones Ca2+ el factor XIa cataliza la ruptura de una unión peptídica en la molécula del factor IX para formar un glucopéptido de 10 KDa y liberar por otro lado al factor IXa.
El factor IX se encuentra ausente en personas con hemofilia tipo B.
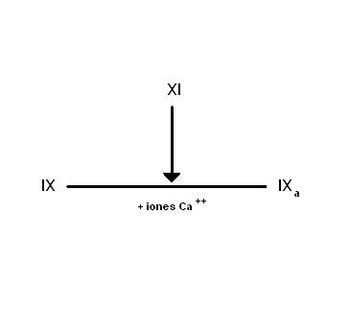 Activación del factor IX
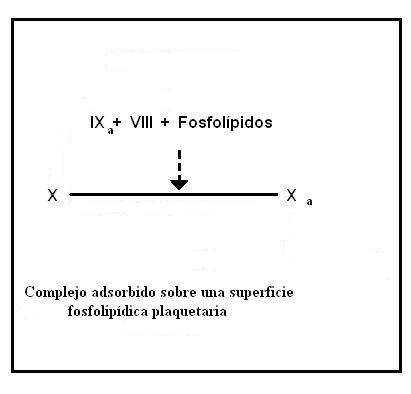
Activación del factor IXFormación del factor Xa
Sobre la membrana de las plaquetas se forma un complejo constituido por los factores IXa, X y VIII.
Los residuos gamma-carboxiglutamato de los factores IXa y X actúan como quelantes del ion Ca2+, permitiendo que estos componentes formen un complejo unido por medio de puentes de iones calcio y ayudando a que el complejo se ancle a los fosfolípidos de membrana.
Primero se unen los factores X y IXa a la membrana y luego se une el VIII.
El factor VIII es en realidad un homorodímero, formado por cuatro cadenas proteicas, cada una codificada por un gen diferente (VIII:C y VIII:R). El componente VIII:C es conocido como "componente antihemofílico" y actúa como cofactor del IXa en la activación del factor X, el componente VIII:R es el que permite la unión del factor VIII al complejo.
La ausencia del componente antihemofílico causa hemofilia A.
El complejo formado por los factores IXa-X-VIII-Fosfolípidos y Ca2+ actúa sobre el factor X para convertirlo en Xa.
En este punto concluye la vía intrínseca.
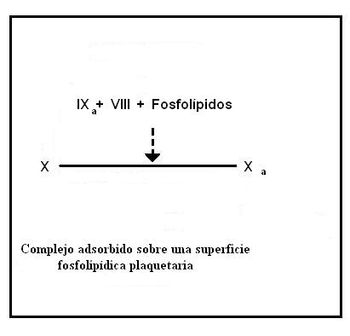 Activación del factor Xu
Activación del factor XuVía extrínseca
Recibió este nombre debido a que fue posible notar desde un primer momento que la iniciación de esta vía requería de factores ajenos a la sangre.
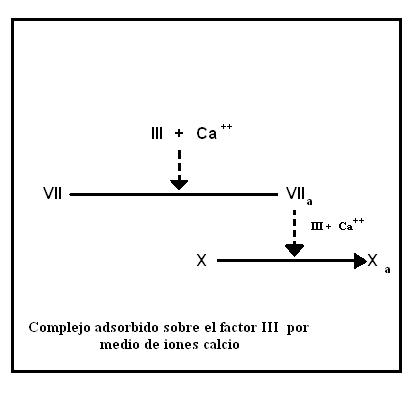
Cuando la sangre entra en contacto con tejidos lesionados o se mezcla con extractos de tejidos, se genera muy rápidamente factor Xa. En este caso la activación de la proenzima X es mediada por un complejo formado por factor VII, Ca2+ y tromboplastina (no confundir con factor III o factor tisular de tromboplastina).
El factor tisular es una lipoproteína sintetizada en el endotelio de los vasos sanguíneos de todos los tejidos, aunque es especialmente abundante en pulmón, cerebro y placenta. El factor tisular se encuentra normalmente "secuestrado" en el interior de las células endoteliales y es secretado en respuesta a una lesión, o bajo el efecto de algunas citoquinas tales como el Factor de Necrosis Tumoral (TNF), InterLeucina 1 (IL-1); o por endotoxinas bacterianas.
La vía extrínseca es muy rápida, se cumple en apenas unos segundos y comprende dos pasos; mientras que la intrínseca insume varios minutos.
Formación del factor VIIa
En primera instancia el factor VII se une a la porción fosfolipídica del factor tisular gracias a sus residuos gamma-carboxiglutamato, utilizando iones Ca2+ como puentes. Este complejo provoca la activación del factor VIIa.
Formación del factor Xa
El complejo VIIa-III-Ca2+ actúa sobre el factor X convirtiéndolo en la proteasa activa Xa. En este punto termina la vía extrínseca y se inicia la vía común
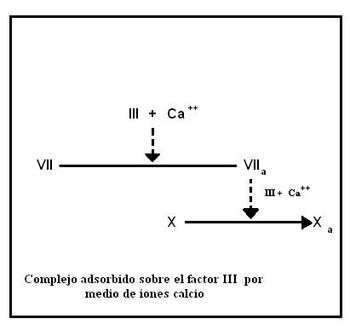 Activación extrínseca
Activación extrínsecaVía común
Llegando al punto en que se activa el factor X, ambas vías confluyen en la llamada vía común.
La vía común termina con la conversión de fibrinógeno en fibrina, y el posterior entrecruzamiento de la misma estabilizando el coágulo.
La vía común implica tres etapas:
Formación de trombina
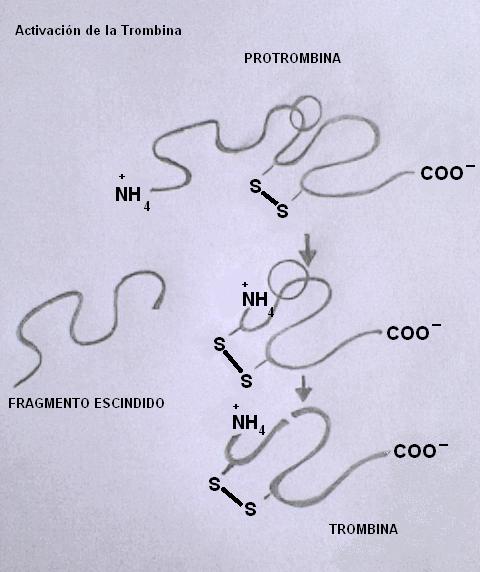
 Representación del mecanismo de activación de la trombina
Representación del mecanismo de activación de la trombinaLa trombina (también llamada factor II a) es una proteasa generada por la ruptura de la cadena proteica de la proenzima protrombina (factor II), una glicoproteína constituida por 582 aminoácidos y con 12 puentes disulfuro intracatenarios.
La trombina se activa luego de que la proteasa Xa hidroliza dos uniones peptídicas de la protrombina. La Xa produce en primer término la escisión de un fragmento de 32 KDa de la región N-terminal de la cadena, cortándola sobre una unión arginina-treonina. En segundo término produce la ruptura de un enlace entre una arginina y una isoleucina; sin embargo estos dos últimos fragmentos permanecen unidos por un puente disulfuro.
La trombina es una serina-proteasa similar a la tripsina, pero mucho más selectiva. Ataca casi de manera exclusiva las uniones arginina con un aminoácido cargado positivamente en sus sustratos.
La conversión de protrombina a trombina debida al factor Xa se acelera notablemente por la formación de un complejo con el factor Va y Ca2+ sobre la superficie de las membranas plaquetarias (fosfolípidos de membrana).
El factor Xa y la protrombina se adsorben sobre la membrana utilizando iones Ca2+ como puentes. El factor Va se une a la protrombina acelerando la reacción.
El factor Va se produce por la acción de la trombina sobre el factor V en un claro ejemplo de una reacción que va acelerándose a medida que progresa (reacción autoacelerada).
Formación de fibrina
El fibrinógeno (factor I) es una glicoproteína compuesta por seis cadenas polipeptídicas: dos A-alfa, dos B-beta y dos gamma; unidas entre sí por puentes disulfuro.
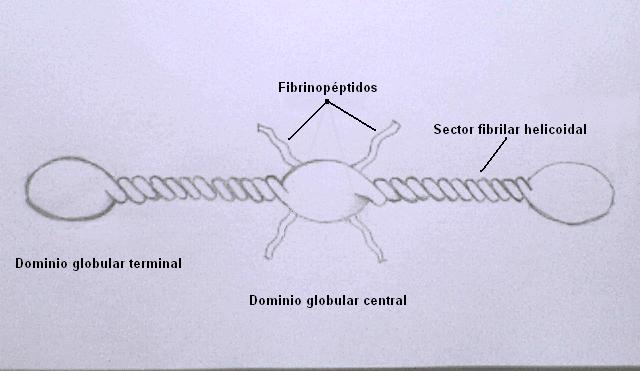
Se trata de una molécula alargada y simétrica formada por tres dominios globulares conectados por segmentos fibrilares.
Cada mitad de la molécula se encuentra formada por tres cadenas (A-alfa, B-beta y gamma) que se enrollan en una triple hélice muy compacta en los sectores fibrilares. Los extremos amino de las seis cadenas se reúnen en el dominio globular central.
En un hecho que parecería muy curioso, los extremos N-terminales de las cadenas A-alfa y B-beta emergen como cabos libres del dominio globular central.
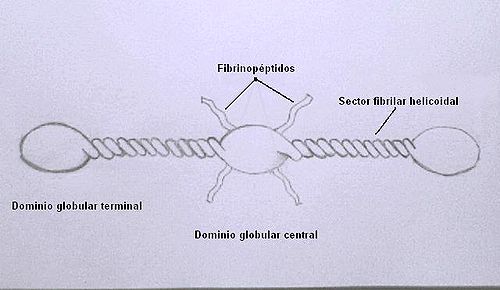 Representación de la molécula de fibrinógeno
Representación de la molécula de fibrinógenoEstas cadenas son muy ricas en aspartato y glutamato, además las cadenas B-beta poseeen en esta región residuos tirosina-O-sulfato formados postraduccionalmente. Estos residuos con una alta tendencia a adquirir carga negativa contribuyen a formar una región central con una muy alta densidad de carga.
Esta región electronegativa central es la responsable de la repulsión entre moléculas de fibrina que las mantiene en solución.
La trombina ataca los enlaces arginina-glicina presentes en estos "cabos libres", separando cuatro péptidos; dos segmentos A de 18 aminoácidos cada uno (provenientes de las cadenas A-alfa), y dos segmentos B de 20 aminoácidos (provenientes de las cadenas B-beta). A estos péptidos se los suele denominar "fibrinopéptidos".
El resto que queda de la molécula es un monómero de fibrina de composición alfa2beta2gamma2.
Al eliminarse los fibinopéptidos desaparecen las fuerzas de repulsión intermoleculares con lo que los monómeros de fibrina tienden a agruparse espontáneamente formando asociaciones altamente ordenadas.
Los monómeros se disponen uno a continuación del otro, cabeza con cabeza en forma de largas hebras. Estas hebras a su vez forman manojos, emparejándose con otras hebras de tal manera que la región central de los monómeros de fibrina de una se encuentra rodeada por las cabezas de los monómeros de fibrina de las otras.
Este emparejamiento se hace posible gracias a interaciones de tipo electrostático y puente hidrógeno entre las regiones centrales de los monómeros de una y las cabezas globulares de otras.
Entrecruzamiento de la fibrina
Los haces paralelos de fibrina polimerizada forman una asociación laxa, que se encuentra en equilibrio con la forma monomérica de la molécula; por lo que sería imposible que cumplieran su papel de formar un coágulo estable sin reforzar esta estructura por medio de enlaces covalentes entre hebras vecinas.
La formación de estos "puentes" covalentes intercatenarios es catalizada por la enzima transglutaminasa (conocida también como factor XIIIa).
La transglutaminidasa cataliza la formación de enlaces amida entre restos glutamina y lisina de hebras próximas entre sí. En la reacción se libera amoníaco en forma de ion amonio (NH4+).
Esta enzima se forma a partir del factor XIII por acción de la trombina.
Regulación y modulación de la cascada
Debido a que la cascada de coagulación consiste en una serie de reacciones que van amplificándose y acelerándose en cada paso, es lógico pensar que debe existir algún mecanismo de regulación; un "freno" a la reacción en cadena; ya que de progresar sin control en pocos minutos podría provocar un taponamiento masivo de los vasos sanguíneos (trombosis diseminada).
Varios mecanismos intervienen en la regulación de la cascada de reacciones:
- El flujo sanguíneo normal, arrastra a los factores activados, diluyendo su acción e impidiéndoles acelerarse. Esta es una de las razones por las cuales cuando existe estasis del flujo sanguíneo se favorece la formación de trombos.
- El hígado actúa como un filtro quitando de la sangre en circulación los factores activados e inactivándolos.
- Existen además algunas proteasas que degradan específicamente a ciertos factores activados, y otras que ejercen acciones inhibitorias sobre factores activos.
Proteína C
La proteína C es una proenzima que se encuentra normalmente en el plasma, y cuya síntesis en el hígado es dependiente de la vitamina K.
Esta proteína es convertida en una proteasa activa por la acción de la trombina.
La proteína Ca actúa específicamente degradando a los factores Va y VIIIa, con lo que limita la proyección de la cascada.
Es interesante notar el triple papel que desempeña la trombina: cataliza la formación de fibrina, activa a la enzima responsable de su entrecruzamiento, y una vez que el proceso de coagulación y estabilización del coágulo está en marcha; ejerce acciones tendientes a limitarlo.
Antitrombina III
La antitrombina III es una glicoproteína de 60 Kda sintetizada en el hígado sin depender de la vitamina K, es considerada la principal inhibidora de la coagulación.
Esta proteína actúa inhibiendo irreversiblemente a varios factores procoagulantes activos, el principal de los cuales es la trombina; aunque también actúa sobre la calicreína y los factores Xa, XIa y XIIa.
La acción de la antitrombina es notablemente aumentada por el heteropolisacárido heparina. La heparina se encuentra en el endotelio de los vasos sanguíneos y en los gránulos de las células cebadas, tiene una poderosa acción anticoagulante ya que facilita la unión de la antitrombina III con los factores procoagulantes activos.
Existen otras anti-proteasas sanguíneas que también ejercen acción anticoagulante aunque menos potente tales como la alfa2 macroglobulina y la alfa1 antitripsina.
Anticoagulantes
Un anticoagulante es, como su nombre lo indica, una sustancia química que retrasa o impide la coagulación de la sangre, ya sea en el interior de un organismo (In Vivo) o en el exterior (In Vitro)
Existen diferentes tipos de anticoagulantes que actúan dificultando o impidiendo alguno de los pasos de la cascada de coagulación.
Existen dos tipos principales de anticoagulantes, los anticoagulantes para uso "In Vitro" y los que tienen empleo "In Vivo", entre estos últimos se encuentran los medicamentos con acción anticoagulante.
En general los anticoagulantes para uso In Vitro actúan como quelantes del ion Ca2+, de manera tal que este no puede participar en la formación de los complejos que activan al factor X, y por lo tanto se interrumpe la cascada de coagulación casi en su inicio.
Los anticoagulantes para uso In Vivo actúan de maneras un poco más complicadas. La adición de algunos agentes quelantes tales como el EDTA entrañan un grave riesgo para la salud del individuo sometido a tratamiento, ya que estos agentes son capaces de acomplejar gran cantidad de iones con alta afinidad, algunos de los cuales desempeñan importantes funciones en el organismo tales como el Cu2+, Fe3+, Zn2+, etc; mientras que otros agentes acomplejantes del calcio tales como el citrato, no tienen gran utilidad ya que son rápidamente metabolizados perdiendo su capacidad anticoagulante.
Entre los anticoagulantes para uso in vivo encontramos sustancias tales como la heparina o los anticoagulantes dicumarínicos.
Para uso In Vitro
- EDTA (C10H16N2O8) o sal disódica, dipotásica o tripotásica del ácido etilendiaminotetraacético. 8 Esta sustancia actúa mediante un efecto quelante sobre el ion calcio (Ca2+, lo que impide la formación de los complejos procoagulantes en los que este ion participa. Este anticoagulante se utiliza fundamentalmente para la realización de recuentos celulares, sobre todo en autoanalizador. Tiene la ventaja de permitir la realización del hematocrito y de frotis sanguíneo hasta dos horas después de la extracción de la muestra. También impide la aglutinación de las plaquetas.
- Heparina Sódica. Heparina de Litio, es un anticoagulante fisiológico que actúa impidiendo que la protrombina se transforme en trombina. Estructuralmente es un mucopolisacárido ácido que posee grupos sulfato. Esta última característica no la hace adecuada para muestras que van a ser examinadas al microscopio luego de tinción, ya que altera notablemente las coloraciónes obtenidas.
- Citrato Trisódico (C6H5O7Na3) actúa impidiendo que el calcio se ionice, evitando así la coagulación. Se utiliza principalmente para realizar pruebas de hemostasia; así como también para medir la velocidad de eritrosedimentación.
- ACD, es un anticoagulante formado por una mezcla de compuestos (Ácido citrico Citrato y Dextrosa en una proporción de 0.9, 2 y 2 g respectivamente en 120 ml de agua destilada) se emplea fundamentalmente en bancos de sangre para conservar las unidades de sangre y para realizar estudios metabólicos eritrocitarios ya que permite una buena conservación de los hematíes.
Anticoagulantes para uso In Vivo (medicamentos anticoagulantes)
Este grupo de anticoagulantes se definen como "medicamentos que impiden la coagulación o la agregación plaquetaria"
Este tipo de medicamentos tienen utilidad en aquellas patologias causadas por un trombo sanguíneo, ya sea para facilitar su disolución (trombolisis) o bien para prevenir que los trombos se repitan.
En este árticulo vamos a centrarnos en aquellos que impiden la cascada de coagulación:
- La heparina alarga el tiempo de coagulación, se administra generalmente mediante inyección subcutánea o endovenosa.
Ya que es un compuesto fisilógico presente en gran cantidad en los mamiferos, comúnmente se utiliza heparina obtenida de pulmón de vaca o de mucosa intestinal de cerdo convenientemente purificada. La potencia difiere según el origen, pero hoy en día vienen estandarizadas en UI, por lo que se pueden comparar solo con este índice.
Comercialmente se obtiene en forma de dos sales (cálcica y sódica) que no guardan demasiada diferencia en su actividad. Las cálcicas se usan preferentemente por vía subcutánea, ya que resultan menos dolorosas, pero por via endovenosa pueden utilizarse ambas. La heparina NUNCA se administra vía intramuscular.
La Heparina se utiliza cuando se precisa de acción anticoagulante rápida y por poco tiempo. En la prevención de trombosis venosas de cirugía se utiliza a bajas dosis, 5.000UI, dos horas antes de la intervención y después cada 12 horas hasta el alta del paciente.
Las heparinas de bajo peso molecular son fragmentos de peso molecular entre 3.500 y 6.000, con ello tiene una vida más larga y aumenta su biodisponibilidad. Tiene una menor inhibición de la agregación plaquetaria. No sustituyen a las heparinas tradicionales sino que en terapias de baja dosis son más cómodas por que se aplican 1 sola vez al día.
En terapias de altas dosis se utilizan las heparinas tradicionales.
- Anticoagulantes dicumarínicos. Reciben este nombre genérico un grupo de compuestos derivados del Dicumarol (un compuesto extraído del trébol dulce) entre los que se encuentran el Acenocumarol (el de uso más frecuente en España, bajo el popular nombre de Sintrom) y la Warfarina. Estos medicamentos presentan la ventaja de poder ser administrados por vía oral y de poseer un efecto prolongado en el tiempo, con gran variabilidad interindividual, por ello necesitan controles periódicos para su ajuste terapéutico.
Todos ellos son inhibidores de la vitamina K (aVK). Debido a que la vitamina K interviene como cofactor enzimático en la síntesis de los factores II,VII,IX y X (concretamente en la gamma-carboxilación de estos); el resultado es que provoca la aparición en sangre, de unas formas inactivas de los mismos denominadas PIVKAs (“Proteins Induced by Vitamin K Antagonists”).
Dada la diferente vida media que presentan los factores de coagulación (el tiempo que pemanecen en sangre antes de ser degradados), por ejemplo el VII comienza a descender en 6 horas pero el II tarda cerca de 70, no se consigue una anticoagulación efectiva hasta el 3º-4º día de tratamiento y el efecto no se estabiliza hasta después de una semana.
Curioso es que la activación de dos inhibidores fisiológicos de la coagulación como son las Proteínas C y S de importancia fundamental(inhiben a los Factores V y VIII activados), también depende de la Vit. K, por lo que los cumarínicos originan una “paradoja bioquímica” anticoagulante-procoagulante.
No obstante, su efecto anticoagulante supera ampliamente al procoagulante, por lo que solo puede tener consecuencias clínicamente significativas en raros casos (Déficits congénitos de Proteína C o S) y de forma transitoria al inicio del tratamiento..
Fibrinólisis
Véase también: CicatrizaciónDespués de que el coágulo se ha establecido, comienza la reparación de los tejidos afectados con el proceso de cicatrización. Para hacer posible esto el coágulo es colonizado por células que formarán nuevos tejidos y en el proceso va siendo degradado.
La degradación de la fibrina (fibrinólisis), componente mayoritaria del coágulo, es catalizada por la enzima plasmina, una serina proteasa que ataca las uniones peptídicas en la región triple hélice de los monómeros de fibrina.
La plasmina se genera a partir del plasminógeno, un precursor inactivo; activándose tanto por la acción de factores intrínsecos (propios de la cascada de coagulación) como extrínsecos, el más importante de los cuales es producido por el endotelio vascular. Se le denomina "activador tisular del plasminógeno" (t-PA).
El gen de este factor ha sido clonado y actualmente se puede obtener la proteína producida por tecnología de ADN recombinante.
Este factor suele utilizarse en clínica para favorecer la disolución de trombos.
Véase también
Enlace externo
Bibliografía
- Ch.4 Haemodynamic diseases. Kumar: Robbins and Cotran Pathologic Basis of Disease 8th Ed. 2009 Saunders (Elsevier)



Wikimedia foundation. 2010.