- Enfermedad de Huntington
-
Enfermedad de Huntington 
George Huntington, quien describió los síntomas de la enfermedad en un artículo de 1872.Clasificación y recursos externos CIE-10 G10 CIE-9 333.4 OMIM 143100 MedlinePlus Información de salud en la enciclopedia MedlinePlus PubMed Buscar en Medline mediante PubMed (en inglés) Sinónimos Corea de Huntington. Baile de San Vito  Aviso médico
Aviso médico 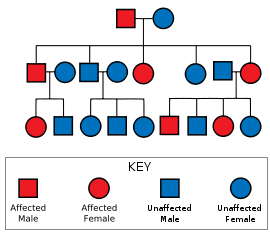 La enfermedad de Huntington tiene herencia autosómica dominante. La probabilidad de cada descendiente de heredar el gen responsable de la mutación es del 50%. La herencia es independiente del sexo, y el gen no se salta generaciones.
La enfermedad de Huntington tiene herencia autosómica dominante. La probabilidad de cada descendiente de heredar el gen responsable de la mutación es del 50%. La herencia es independiente del sexo, y el gen no se salta generaciones.
La enfermedad de Huntington (llamada también corea[1] de Huntington y conocida antiguamente como baile de San Vito o mal de San Vito, al igual que otras coreas como la corea de Sydenham) es un trastorno genético hereditario cuya consideración clínica se puede resumir en que es un trastorno neuropsiquiátrico. Sus síntomas suelen aparecer hacia la mitad de la vida de la persona que lo padece (unos 30 o 50 años de media) aunque pueden aparecer antes y los pacientes muestran degeneración neuronal constante, progresiva e ininterrumpida hasta el final de la enfermedad que suele coincidir con el final de su vida por demencia y muerte o suicidio. Esta enfermedad genética presenta una herencia autosómica dominante, lo cual significa que cualquier niño en una familia en la cual uno de los progenitores esté afectado, tiene un 50% de probabilidades de heredar la mutación que causa la enfermedad.
El padecimiento de la enfermedad puede seguir caminos muy diferentes, incluso entre hermanos y parientes próximos. Esto se debe a que, junto a la mutación específica del gen de la huntingtina,[2] intervienen además otros factores hereditarios.
La enfermedad produce alteración cognoscitiva, psiquiátrica y motora, de progresión muy lenta, durante un periodo de 15 a 20 años. El rasgo externo más asociado a la enfermedad es el movimiento exagerado de las extremidades (movimientos coréicos) y la aparición de muecas repentinas. Además, se hace progresivamente difícil el hablar y el tragar. En las etapas finales de la enfermedad, la duración de los movimientos se alarga, manteniendo los miembros en posiciones complicadas y dolorosas durante un tiempo que puede prolongarse hasta horas.
No obstante, los trastornos psíquicos graves, que anteceden normalmente a los musculares, son los rasgos característicos de la enfermedad. Ésta puede desencadenar episodios depresivos reiterados con repercusiones negativas en el entorno de allegados. Las facultades cognitivas disminuyen, así como la memoria, y la capacidad de concentración empeora. La enfermedad termina en una demencia fuerte, que puede conllevar deseos de suicidio.
En 1872, el médico George Huntington, observó por primera vez esta enfermedad en una familia americana de ascendencia inglesa y le dio el nombre de «enfermedad de Huntington». El nombre alternativo de «corea» viene porque entre sus síntomas visibles encontramos movimientos coréicos, es decir, movimientos involuntarios y bruscos de las extremidades. Se cree que los orígenes debieron ser en el noroeste europeo y que desde allí se extendió al resto del mundo, especialmente a América donde encontramos tasas elevadas de esta afección. En 1933 se descubrió que el desencadenante de la enfermedad era una mutación genética localizada posteriormente en el cromosoma 4,[3] lo cual se publicó en la revista Nature en 1982 por el equipo de genética de la Facultad de Medicina de la Universidad Harvard, Boston.
La población más grande conocida con la Enfermedad de Huntington se encuentra en la región de la Costa Occidental del Lago de Maracaibo - Estado Zulia, en Venezuela, y se estima que llegó allí a principios del siglo XIX y que, como consecuencia de un efecto fundador, se ha mantenido y hay muchos miembros de la población que la padecen y los que no, tienen un alto riesgo de padecerla. Gracias a esta población, y a las muestras para análisis que cedieron sus miembros, en 1983, varios equipos de investigación entre los que cabe destacar el de J.F. Gusella, descubrieron mediante técnicas de ligamiento la localización exacta de esta enfermedad en el genoma humano. El gen responsable es el llamado «gen de la huntingtina» que encontramos cerca del telómero del brazo corto del cromosoma 4.
Se calcula (2006) que en toda Europa hay unos 45.000 afectados. En Norteamérica, unos 30.000.
Tras llegar a la mayoría de edad, cualquier individuo puede hacerse un examen predictivo y obtener así la seguridad o no de su presencia con años e incluso decenios de anticipación a sus primeros síntomas. El examen genético es infalible pues todo portador de esa mutación genética se convertirá, antes o después, en víctima de la enfermedad.
Actualmente, existe también el diagnóstico preimplantacional: en una fertilización in vitro, se analiza cuál de los embriones que se han comenzado a desarrollar presenta la enfermedad, y cuál no, implantando únicamente el sano, de tal manera que el hijo deseado no estará afectado por esta enfermedad.
Contenido
Etiología
La enfermedad se produce mediante un único factor hereditario. El defecto genético se encuentra a nivel del cromosoma 4. Afecta a una proteína de función desconocida y expresión en numerosos tejidos, llamada Huntingtina. El defecto se debe a una expansión de tripletes CAG que codifican la síntesis de la glutamina. En la secuencia original hay 34 repeticiones, y en la enfermedad, más de 40. Aunque todavía no están establecidas completamente las bases fisiopatológicas de la enfermedad, se cree que esas «colas adicionales de glutamina», hacen que las proteínas interaccionen entre sí de manera hidrofóbica y se facilite la formación de precipitados y acúmulos proteicos, especialmente en el cerebro.
El número de repeticiones está relacionado en proporción directa con la gravedad de los síntomas y es inversamente proporcional a la edad de presentación. En este tipo de enfermedades por expansión de tripletes, es frecuente que un ligero incremento en el número de repeticiones no produzca la enfermedad, pero que ese incremento se transmita a las generaciones futuras, produciéndose, en cada gametogénesis, un incremento en el número de repeticiones, hasta finalmente inducir la enfermedad. En el momento en que está establecida, la herencia es autosómica dominante (es decir, cada descendiente tiene un 50% de posibilidades de heredar la enfermedad). Una de las características de este tipo de enfermedades de expansión de tripletes es la anticipación génica, es decir, conforme van pasando las generaciones, el número de repeticiones se amplía, y eso hace que la enfermedad se manifieste antes y más agresivamente en las generaciones futuras. Además del fenómeno de anticipación génica, también es algo habitual en este tipo de enfermedades el fenómeno de impronta genética. En el caso de la Enfermedad de Huntington, la impronta genética es de tipo paterno, lo que se traduce en que las modificaciones en la expresión del gen de la huntingtina se producen a través de la línea germinal paterna (en términos de afección genética, la elongación de la región donde se localizan las repeticiones de trinucleótidos se produce en la meiosis paterna, en la formación de los gametos).
Enfermedad de Huntington juvenil
Normalmente se refiere a los pacientes que manifiestan la enfermedad antes de los 20 años aunque se consideran más en esta denominación a los casos de enfermos menores de 12 años. En esta modalidad se incluyen menos del 10% de los casos de enfermos de la enfermedad de Huntington que se conocen, lo que quiere decir que no es la predominante. En la mayoría de los casos, al igual que en la variante normal, se asocia con transmisión paterna y número de repeticiones de los tripletes CAG muy elevados ( de 60 en adelante). Los síntomas más notables son la rigidez, contracción y agarrotamiento de los músculos. El primero en describirla fue Hoffmann en 1888, que identificó a dos niñas de 4 y 10 años con algunos de los síntomas como rigidez, hipoquinesia y agarrotamiento muscular.
Un ejemplo destacable es, por ejemplo, el del grupo de investigación de Nahhas (2005) que investigó el caso de una niña con un historial de la enfermedad en la rama materna de su familia. Presentó los primeros síntomas a los 3 años y murió a los 7 por complicaciones clínicas de la Corea de Huntington. La madre de la niña presentó sus primeros síntomas a los 18 años de edad. El análisis de las muestras de ambas, desveló que la madre tenía 70 repeticiones de los tripletes, mientras que la niña presentó aproximadamente 130 repeticiones. Con este caso, el grupo de Nahhas manifestó que se trataba del mayor caso confirmado molecularmente de expansión de tripletes CAG en transmisión materna, demostrándose que las expansiones grandes también se pueden dar (aunque sean menos frecuentes) en la línea germinal materna.
El síndrome de Westphal es también la manifestación de la enfermedad de Huntington a edades tempranas. En concreto, este síndrome engloba los casos que se den en pacientes de menos de 20 años de edad. Por lo demás es bastante similar a la Corea de Huntington juvenil.
En cualquier caso, los síntomas son similares a la enfermedad de Huntington que se ve en adultos, aunque en muchos de los casos se ven agravados por la juventud de los pacientes y la dificultad de controlar sus problemas mentales y físicos.
Incidencia
Se estima que la incidencia media está en 4 u 8 afectados por cada 100.000 personas. A pesar de ello, se sabe que hay grandes diferencias entre las poblaciones humanas, siendo las poblaciones asiáticas, por ejemplo, menos propensos, mientras que en Reino Unido la incidencia aumenta bruscamente.
Diagnóstico
Sospecha por la clínica y confirmación por diagnóstico molecular. RM, TAC, TEP o pruebas neuropsiquiátricas pueden ser inespecíficas o reflejar la atrofia cerebral en la cabeza del núcleo caudado y de la corteza cerebral, y dilatación ventricular. Puede ser normal en enfermedad precoz. Hoy en día, se aplican métodos para detectar las mutaciones específicas (en este caso, contar el número de repeticiones). Lleva aparejado siempre el consejo genético.
El diagnóstico diferencial debe hacerse con esquizofrenia, corea familiar benigno, ataxias hereditarias, acantocitosis neural, enfermedad de Alzheimer, enfermedad de Pick o enfermedad de Creutzfeldt-Jakob.
Unos pocos individuos desarrollan la EH >55 años: EH de inicio tardío. Prevalencia del 25%. Diagnóstico difícil. Progresión lenta. Los síntomas pueden enmascararse por otros problemas de salud. Signos de depresión antes que cólera o irritabilidad. Pueden conservar un control marcado de sus funciones intelectuales: memoria; razonamiento; resolución de problemas. Muerte por causas no relacionadas con EH. Técnicas de genética molecular (PCR): confirmación de la enfermedad y diagnóstico presintomático. Individuos sin EH tienen 28 o menos CAG repetidas. Los individuos con EH poseen más de 40 repeticiones. Un pequeño porcentaje tienen un nº dentro de la región bordeline.
Clasificación de las repeticiones de nucleótidos y el estado de la enfermedad de cada persona dependiendo de la cantidad de repeticiones del trinucleótido CAG.[4] Nº de repeticiones CAG Clasificación Resultado <28 rango normal No desarrollo de EH. 29–34 intermedio El individuo no desarrolla la EH, pero la siguiente generación está en riesgo. 35–39 penetrancia reducida Algunos, pero no todos, desarrolla la EH. La siguiente generación está en riesgo. >39 penetrancia total Desarrollo la EH. Al haberse descubierto la mutación, ya es posible determinar si los casos sin historia familiar y con edad de comienzo y manifestaciones clínicas compatibles son en realidad casos de enfermedad de Huntington.
Test presintomático
Identificación de personas portadoras del gen EH, antes de aparición de síntomas. Más barato, más sencillo y más exacto. Usa la longitud de repetición de CAG para detectar la presencia de la mutación de EH en sangre. Puede requerir una muestra de ADN de un familiar afectado muy cercano, preferiblemente un padre. No se recomienda en <18 años, para proteger los intereses, incluyendo la confidencialidad, excepto si existe una razón médica convincente (presencia de síntomas). Se debe informar al paciente y a la familia sobre los riesgos relativos médicos y psicosociales y los beneficios de conocer el estado de portador del gen de EH.
Realización
Los programas constan:
- examen neurológico: Si un individuo muestra algún síntoma aunque sea ligero, de EH, será diagnosticado de EH, incluso antes del test genético.
- consejo pretest: El individuo recibirá información sobre:
· EH, · su nivel de riesgo, · sobre el procedimiento, exactitud, limitaciones y resultados de la prueba.
- seguimiento
Indicaciones
- Niño < 10 años que presenten estos criterios:
Historia familiar de EH ( normalmente en el padre ) y 2 o más de los siguientes:
- Fracaso
- Crisis convulsivas
- Disfunción oral motora
- Rigidez
- Trastornos de la marcha
Si no cumplen estos criterios clínicos, se propone:
- Tratamiento sintomático.
- Posibilidad de realización del test posteriormente, si apareciera algún síntoma sugestivo de EH.
- Jóvenes entre 10-20 años; no hay criterios establecidos, pero se debe tener en cuenta:
- Adolescentes con riesgo de presentar EH, con alteraciones del comportamiento o síntomas motores, pueden o no presentar, EH.
- Adolescentes sin Hª familiar (paterna) de EH, es improbable que desarrollen la enfermedad.
Test prenatal
Algunos padres desean conocer el riesgo del feto, pero no el suyo propio. Se usan marcadores ligados al ADN, en vez de un test genético directo. El test no busca el gen EH en el padre. Indica si el feto ha adquirido el cromosoma 4 de un abuelo afectado o de uno que no lo esté, en el marco de una familia con EH. Si la prueba demuestra que el feto ha heredado el crom 4 del abuelo afectado, los padres sabrán que el riesgo es el mismo que el del padre (50-50). Si el test muestra que el feto ha adquirido el crom. 4 de un abuelo no afectado, el riesgo es muy bajo (<1%). Alternativa: Fertilización con screening pre-implantación. Los embriones son estudiados para determinar cuál es portador de la mutación EH. Los embriones sin mutación son implantados en el útero de la mujer.
Implicaciones ético-sociales y consejo genético
Actualmente y más en concreto en esta enfermedad, se puede determinar la manifestación futura de la misma mucho antes de que empiecen los primeros síntomas en un paciente y también el riesgo al que están expuestos los familiares. Esta determinación requiere un análisis de marcadores de ADN cercanos al gen de la huntingtina pero las muestras deben ser también de miembros de la familia del paciente. Además hay que añadir que un diagnóstico presintomático se puede traducir en una sentencia de muerte ya que no hay cura a la Enfermedad de Huntington. Todo esto supone una serie de problemas ético sociales que tienen que tener en cuenta los médicos y consejeros genéticos. Por ejemplo, si es una prueba presintomática, el paciente debe dar su consentimiento para que se informe a sus parientes del resultado y las consecuencias que les afectan; en el sentido contrario de la cadena de acontecimientos, los parientes deben estar de acuerdo en ceder sus muestras de sangre y cada persona relacionada debe estar informada acerca de las consecuencias que pueda tener su decisión y tiene total derecho a tomar una decisión propia. De esta forma cada uno tiene derecho a no conocer su situación pero esto choca con el derecho de los familiares a conocer esa información. Esto supone un problema con la confidencialidad (obligada en medicina) en pruebas de personas emparentadas y de hecho, en estudios que se han realizado, el 50% de los familiares de pacientes con Corea de Huntington se han negado a conocer su estado con respecto al gen de la huntingtina.
Clínica
La clínica, como ya hemos mencionado, es de presentación tardía (4ª-5ª década de la vida), y se caracteriza por movimientos coréicos o corea (movimientos espasmódicos e involuntarios, amplios y bruscos de las extremidades, que dificultan incluso la marcha), síntomas psiquiátricos (problemas afectivos y cambio de personalidad, irritabildiad, agresividad, brotes psicóticos, deseo de suicidio) y una degeneración neurológica progresiva que llega a conducir a la demencia. También se caracteriza por parkinsonismo, pérdida de expresión facial (por su combinación con la corea, se ha llamado a esta enfermedad de "máscara en movimiento"), e incluso trastornos oculares (movimientos sacádicos, alteraciones en el parpadeo).
El orden de presentación de los síntomas es casi siempre el de los no-motores antes que los motores, siendo los primeros los psiquiátricos (los oculares pueden ser un signo precoz), luego movimientos nerviosos ("inquietos"), que dan paso al cuadro general, y los más avanzados son los debidos al deterioro neurológico (sobre todo a nivel cognitivo, del lenguaje, dificultad en la deglución, etc.). La muerte (de media 15 años después del inicio de la enfermedad) suele deberse a una neumonía por aspiración u otra complicación médica, cuando no se trata de casos de suicidios.
La alteración cognitiva de la EH junto con otras patologías neurológicas de origen subcortical, por ej. enfermedad Parkinson, representa un importante modelo humano de la disfunción cognitiva de los ganglios basales.
Los déficits de memoria y aprendizaje son el trastorno cognitivo más característico y precoz de esta enfermedad. Probablemente estén presentes durante varios años previos a la aparición de los movimientos coreicos. Los déficits mnésicos se relacionan con problemas de la recuperación de la información, más que con problemas de almacenamiento del material aprendido, como consecuencia de un deterioro del neoestriado.
Los principales déficits neuropsicológicos se centran en una alteración de la atención y de la capacidad de concentración, pensamiento enlentecido, incapacidad para operar con un conocimiento adquirido, disminución de capacidad de aprendizaje visual y verbal, así como su posterior recuperación, falta de planificación y ordenación secuencial, para terminar en un déficit de capacidad de solución de problemas y formación de conceptos. Los aspectos motores, visuoespaciales, memoria visual inmediata y remota, y las disfunciones frontales nos permiten diferenciar entre pacientes con un leve y moderado deterioro de la capacidad funcional.
La duración de los síntomas neurológicos o de los síntomas psiquiátricos son pobres indicadores predictivos del estado cognitivo y funcional del paciente. No obstante, el curso gradual y cognitivo de los déficits de las funciones cognitivas es paralelo al deterioro motor.
Algunos trabajos han sugerido la existencia de tres grupos evolutivos distintos en función del deterioro neuropsicológico:
- 1. Signos de deterioro subcortical: disartria, bradipsiquia, bradicinesia y pseudoalteración de la memoria. Estas alteraciones responden a alteraciones de los gg. basales propias de los primeros estadios de la enfermedad.
- 2. Signos de deterioro subcortical más signos de disfunción frontal: alteraciones del cálculo mental y escrito, adinamia verbal, cierto grado de agrafía, alteraciones de la secuenciación motora y de la capacidad de inhibición.
- 3. Signos afaso-apraxo-agnósicos y una mayor afectación de las funciones motoras y premotoras, que podría reflejar una generalización de la degeneración que afectaría al córtex cerebral, propia de las fases avanzadas de la enfermedad.
A medida que progresa la enfermedad se hace más evidente el deterioro de las funciones intelectuales, especialmente del factor manipulativo, y se observan diferencias intercociente entre el factor verbal y manipulativo. Se aprecia un declive del coeficiente de inteligencia (CI), aunque difícilmente se encuentra un CI < 70 en sujetos de menos de 10 años de evolución. No obstante el CI global suele ser < 100.
Las funciones lingüísticas se hallan preservadas en estadios iniciales de la enfermedad.
- 1. En fases iniciales la neurodegeneración afecta a las zonas anteromediales del caudado y dorsales del putamen. La primera posee conexiones con el córtex dorsolateral frontal. La segunda recibe aferencias del córtex premotor. Se detectan alteraciones de la articulación (disartria hipercinética). En el lenguaje espontáneo se observa: escasos errores lingüísticos, pocas parafasias semánticas, pocos errores paragramáticos o agramáticos, repetición preservada, una preservación de la denominación por confrontación visual, ausencia de déficits de comprensión y alteración de la fluidez verbal.
- 2. En estadios intermedios se presenta reducción del número de palabras y de la fluidez verbal, y una alteración de la agilidad articulatoria. Alteración de la repetición, ligera disminución de la complejidad sintáctica, reducción de la línea melódica y de la longitud de la frase, alteración de la forma gramatical sin objetivarse agramatismo, incremento del número de parafasias semánticas con leve dificultad para encontrar palabras en el lenguaje espontáneo y una moderada alteración de la comprensión. La escritura también está afectada.
- 3. En la enfermedad evolucionada se afectan zonas más posteriores del caudado y las porciones del putamen que reciben proyecciones directas de la circunvolución temporal superior. Alteraciones lingüísticas con características de una afasia de Wernicke. Marcada reducción de la fluidez verbal y de la complejidad sintáctica de las oraciones en su lenguaje espontáneo, presencia de estereotipias verbales o perseveraciones sin ecolalia, alteración de la repetición, marcados déficits en la capacidad de comprensión, e importante incremento en la producción de parafasias semánticas. La escritura aparece disgráfica, se pueden presentar sacudidas imprevisibles, la sintaxis se vuelve incompleta y se dan omisiones o sustituciones léxicas. Finalmente, se presenta una marcada alteración de la lectura caracterizada con múltiples autocorrecciones, sustituciones, adicción de letras y palabras, omisiones e indecisión en la producción lectora.
Las alteraciones en el procesamiento visuoespacial son evidentes en los pacientes con EH y se distinguen de las alteraciones visuoespaciales que están presentes en otras demencias. Se manifiestan tanto en estadios iniciales como en los de moderado deterioro cognitivo. Muestran alteraciones del procesamiento visuoespacial general, de la integración perceptivo-motora, de la manipulación de la información espacial, de la rotación mental espacial, del sentido de la dirección, de la discriminación visuoespacial y de la percepción espacial egocéntrica, con una preservación del juicio visuoespacial. Algunos individuos son incapaces de reconocer caras.
Genética
Descubrimiento del gen
En 1983, seis grupos de investigación, entre los que destacó el de James Gussella, consiguieron aislar el gen de la huntingtina. Esta enfermedad es una de las primeras en las que los métodos de genética molecular ayudaron al descubrimiento de un marcador de ADN ligado al gen, lo que permite realizar diagnósticos presintomáticos e incluso prenatales de algunos individuos. La investigación de Gussella y cols. fue una de las primeras en tener éxito en el uso de RFLPs (Polimorfismos en Longitud para Fragmentos de Restricción) para demostrar ligamiento y fue con la Enfermedad de Huntington.

Al principio de la investigación se intentó identificar el ligamiento con marcadores protéicos en suero, pero no funcionó. Entonces, se encontró una población considerablemente amplia que padecía la enfermedad en la mayoría de sus miembros: la población venezolana de Maracaibo. Gracias a las muestras de ADN cedidas por sus miembros y a la suerte (en esta época había pocos marcadores polimórficos de ADN disponibles) se logró encontrar un marcador. La sonda G8, que fue una de las que se utilizó, encontró dos polimorfismos con la enzima de restricción Hind III. Se ha demostrado la relación entre la enfermedad y el locus marcador, y se ha establecido que la G8 mapea de 3 a 5 cM del gen de la huntingtina.
Gracias a este gran descubrimiento en la eficacia de los RFLPs en mapeo de genes de enfermedades, cada día son más los trastornos monogenéticos que se consiguen localizar en una región cromosómica concreta. La importancia de esto radica en que da la oportunidad, por primera vez en muchos casos, de hacer diagnósticos presintomáticos o prenatales en los individuos en riesgo.
Gen de la Huntingtina
El gen de la huntingtina se mapeó físicamente en la banda más distal del cromosoma 4 humano, determinando su localización en esa región. Se trata de una zona bastante complicada de estudiar porque está muy próxima al telómero del brazo corto del cromosoma. El gen se sitúa exactamente en 4p16.3. Esta región es de aproximadamente 1000 Kb y tiene un contenido génico potencial de unos 5000 genes de los cuales se sospecha que 27 están relacionados con enfermedades. El gen de la huntingtina produce una proteína llamada huntingtina cuya función aún es desconocida.
Tras conseguir aislar el gen, se encontró una conexión con otras tantas enfermedades hereditarias: el mecanismo de mutación en todas ellas es la expansión de repeticiones de trinucleótidos. En la Enfermedad de Huntington, el aumento del número de tripletes en una posición concreta en el gen, es lo que diferencia al gen mutado de la huntingtina del gen normal. En el extremo 5' del gen es donde se localiza el grupo de tripletes que se repite (CAG). Los tripletes se sitúan en el primer exón y codifican para el aminoácido glutamina. La proteína normal es polimórfica para un segmento de poliglutamina. Las repeticiones de tripletes varían normalmente entre 8 y 35, siendo 35 lo que se considera un umbral y a partir de aquí se considera enfermedad. 35 es un umbral inexacto ya que hay casos excepcionales en los que los niveles mínimos de unos enfermos se superponen con los niveles máximos de la enfermedad. Antes del umbral, podemos ver casos de números intermedios de repeticiones que pueden suponer una predisposición a padecer la enfermedad (premutación) en la siguiente generación.
Anatomía patológica
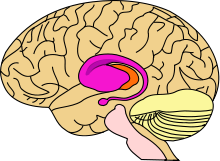 Área del cerebro dañada por EH – cerebro caudado y putamen (en rosa)
Área del cerebro dañada por EH – cerebro caudado y putamen (en rosa)Desde el punto de vista anatonomopatólogo, se sabe que lo que sucede es una degeneración neuronal y que más en concreto comienza en las neuronas medianas (en las neuronas espinosas medias), conservándose las neuronas grandes. Se producen daños graves y visibles en el cuerpo estriado (en el núcleo lenticular y el núcleo caudado) del cerebro, es decir, atrofia del cerebro en las zonas parietales, frontal y en el tálamo y el putamen principalmente. La corteza cerebral se mantiene más o menos bien hasta que la enfermedad está bastante avanzada y en lo que se refiere a la corteza cerebelosa, no se ha visto que sufra daño morfológico alguno. Los movimientos involuntarios ponen de manifiesto la localización de daños en el sistema extrapiramidal. En resumen se puede decir que se produce una atrofia cerebral caracterizada por muerte neuronal y gliosis.
La mutación del gen y su mecanismo
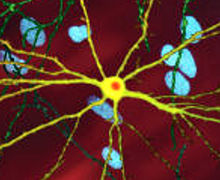 Una imagen de microscopía de una neurona con inclusiones (teñidas de naranja) causadas por EH, ancho de la imagen 250 µm
Una imagen de microscopía de una neurona con inclusiones (teñidas de naranja) causadas por EH, ancho de la imagen 250 µmEl gen de la huntingtina se expresa en diversos tejidos predominando en el cerebro la huntingtina normal. La localización de la proteína en el organismo se ha concretado mediante inmunohistoquímica (con anticuerpos monoclonales) y se ha detectado en el citoplasma neuronal, el pericarion, las fibras nerviosas y las terminaciones sinápticas. Por esto mismo, aunque se desconoce la función de la proteína normal, se demuestra que debe ser una proteína estructural de terminaciones nerviosas y no un regulador transcripcional. Se ha visto que los enfermos de Huntington, producen tanto la proteína mutada como la normal y la conclusión a la que se ha llegado es que la dominancia se traduce en una ganancia de función por parte de la proteína y por eso apenas hay variaciones clínicas entre homocigotos y heterocigotos. La función que adquiere la proteína mutada guarda relación con la atrofia cerebral y con la degeneración de las neuronas espinosas medianas, primero en el putamen y luego en el encéfalo en general. Los últimos estudios realizados al respecto demuestran que el extremo aminoterminal con poliglutamina de la proteína mutada, es reconocido como un plegamiento anormal y es atacada por caspasas específicas para eliminar ese extremo. Esto provocaría que los restos se agruparan y se fueran acumulando. Estas acumulaciones producirían problemas y dificultarían la regulación en el núcleo de la célula empeorando con la edad. En resumen, la acumulación resultaría tóxica para la célula interrumpiendo la actividad de degradación de proteínas de las células que ya decrece con la edad y tendría un efecto acumulativo.
Se sabe que la mutación del gen de la huntingtina no ha producido otras variaciones, es decir que otras mutaciones en el gen de la huntingtina son raras por lo que se cree que el surgimiento de la mutación conocida y por tanto de la enfermedad, se debió a un avance progresivo de la elongación de la zona de repeticiones hasta superarse el umbral. La mutación se ha mantenido por la transmisión de generación en generación, en la generalidad de los casos.
Tratamiento
No existe tratamiento que cure la enfermedad ni que impida la progresión. La medicación disponible se limita a contrarrestar la sintomatología, así como la cirugía cerebral puede disminuir considerablemente el progreso de la enfermedad.
Contra los trastornos motores se recetan neurolépticos tipo tiaprida y tetrabenazina, que aunque orientados en principios a la psicosis esquizofrénica, limitan secundariamente los movimientos de los pacientes. También se usan bloqueantes de dopamina (fenotiacina, haloperidol) y otros medicamentos (amantidina, reserpina).
Para los trastornos psíquicos se utilizan antidepresivos, sedantes y neurolépticos antipsicóticos.
Además, existe un tratamiento de rehabilitación, psiquiátrico y psicológico, nutricional, y sobre todo, de apoyo social.
Si se inicia el tratamiento farmacológico, las dosis de inicio de neurolépticos deberán ser bajas, por ejemplo, 0,5-1 mg/día de haloperidol o flufenacina. Las dosis pueden aumentarse gradualmente con incrementos mínimos (1 mg/día) hasta que se alivien los síntomas. Con concentraciones en suero de 2-5 ng/ml aparece mejoría, y corresponden a una dosis diaria de 1,5-10 mg/día. Dosis >10 mg/día de haloperidol producen solo pequeños o ningún beneficio que dosis menores. Si los pacientes presentan rigidez, acatisia o reacciones distónicas por el haloperidol o la flufenacina, fármacos menos potentes como la tioridacina pueden ser mejor tolerados. Sin embargo, los neurolépticos menos potentes son más sedantes, más anticolinérgicos y pueden causar más hipotensión postural que los más potentes.
Se ha utilizado una gran variedad de fármacos para el control de la rigidez, espasticidad, y distonías pero sin mucho éxito. Una excepción es la toxina botulínica (IM), que se ha usado con bastante éxito en la distonía cervical en la EH juvenil.
Clonacepam y valproato se han usado para las miclonías; y el valproato puede ser particularmente efectivo en pacientes jóvenes con EH con epilepsia más que en aquellos que presentan crisis primarias generalizadas. Como con otras crisis compulsivas, las secundarias a la EH deben ser evaluadas con un EEG.
Tratamiento de las alteraciones psiquiátricas
La mayoría de las depresiones en la EH responden al tratamiento de la depresión idiopática. En general, la depresión de la EH está mal diagnosticada y mal tratada. A pesar de que no existen estudios controlados, pueden ser efectivos tanto los antidepresivos tricíclicos como los inhibidores selectivos de la recaptación de serotonina (ISRS). Los IMAOs también han sido utilizados con éxito. Los ISRS son más cómodos de manejar porque no requieren monitorización de niveles sanguíneos, tiene un muy bajo potencial de mortalidad en casos de sobredosis y presentan a menudo el beneficio adicional de mejorar los síntomas de irritabilidad y agresión. Con estos fármacos los pacientes pueden desarrollar acatisia y un empeoramiento de su insomnio, incluso en raros casos incrementar las disquinesias. Entre los antidepresivos tricíclicos se prefiere la nortriptilina. Su baja actividad anticolinérgica conlleva una menor sequedad de boca, menos estreñimiento y menos visión borrosa. Su baja actividad alfa-bloqueante, minimiza la hipotensión ortostática.
Los pacientes con EH pueden no necesitan tratamiento farmacológico en temporadas de buena evolución si son breves y no hay asociados comportamientos peligrosos. La carbamazepina o el valproato sódico son el tratamiento inicial de elección, comenzando con pequeñas dosis, incrementándola gradualmente hasta que aparezca respuesta.
En el tratamiento de la irritabilidad se ha tenido éxito con los ISRS y la carbamazepina.
Las alteraciones sexuales en EH, particularmente agresividad hipersexual, pueden ser tratadas con antiandrógenos.
Las alteraciones obsesivas compulsivas en la EH pueden ser tratadas con los fármacos estándar en el tratamiento de las obsesiones, tales como ISRS y clomipramina.
Investigación terapéutica
Investigación farmacológica
La búsqueda de los investigadores se centra en el descubrimiento de sustancias que retarden, si no impidan, el proceso de degeneración neuronal. Un ejemplo de las mismas serían los antagonistas de los receptores glutamínicos, que dificultan la liberación del trasmisor glutamato.
Un antibiótico, la minociclina (usado contra el acné), resulta efectivo a la hora de inhibir las caspasas, que son las enzimas que desencadenan la necrosis de las células nerviosas (se ha demostrado ya efectiva en ratones).
Para impedir la degradación de la proteína huntingtina se está investigando en sustancias como la trehalosa (un azúcar procedente de plantas del desierto) que permitirían retardar el comienzo de la enfermedad.
Se ha recurrido, también, a intervenir en el metabolismo energético alterado de la célula, utilizando para ello sustancias del cuerpo del paciente (coenzima Q, antioxidante, y creatina, depósito de energía). Los ensayos en animales resultan esperanzadores.
El uso de antitumorales es otra de la líneas de investigación terapéutica; el fenilbutirato podría actuar poniendo de nuevo en funcionamiento la síntesis proteica, que se ve alterada por la huntingtina modificada.
Ensayos de terapia genética
El principal logro en este terreno ha sido el lograr impedir en ratones la expresión del gen de la huntingtina modificada, inyectando en el cerebro pequeños fragmentos de ARN que coincidían con el ARN portador de la información para elaborar la proteína alterada patológicamente y bloquearla.
En cuanto a las células madre, se ha implantado en pacientes células madre neuronales procedentes de fetos abortados con la esperanza de que lleguen a sustituir a la afectadas. Los resultados han sido dispares de momento.
Curiosidades e historia
- En el pasado, no se diagnosticaba como tal (de ahí el problema de que muchos individuos no sean conscientes de sus antecedentes familiares).
- En el medievo, se conocía a esta y otras enfermedades similares (corea de Sydenham) como "El baile de San Vito" pues las personas aquejadas de los movimientos espasmódicos característicos que dificultan la marcha peregrinaban a la capilla de San Vito, construida en Ulm (Alemania), esperando que el santo los curara.
- Existen comunidades enteras en el continente americano donde la enfermedad es un mal endémico (debido a que la traían consigo los primeros colonizadores); en la región de la Costa Occidental del Lago de Maracaibo en Venezuela el número de casos supera hasta diez veces el promedio mundial.
- En la serie de Fox House M.D. ., desde la cuarta temporada se sabe que la doctora internista Remy Hadley (mejor conocida como 13), padece esta enfermedad.
Notas
- ↑ La palabra viene del griego choreia, danza, y hace referencia al síntoma que originalmente servía para denominar la enfermedad: los movimientos danzarines exagerados de las extremidades.
- ↑ La huntingtina es una proteína imprescindible para el desarrollo embrionario de los vertebrados. Además, interviene en la comunicación entre las células nerviosas.
- ↑ Se trata de uno de los 22 cromosomas no ligados al sexo, haciendo a las mujeres y los hombres tener el mismo riesgo de adquirir la enfermedad. El gen normal tiene tres bases de ADN, compuesta por la secuencia CAG. La mutación genética consiste en un segmento de ADN inestable, donde varios pares de bases se repiten docenas de veces. La expansión del trinucleótido repetido - (CAG)n - origina la enfermedad de Huntington por su efecto sobre la expresión o estructura de la proteína codificada por el gen 1T15 (la huntingtina).
- ↑ Walker FO (January 2007). «Huntington's disease». Lancet 369 (9557): pp. 218–28. doi:. PMID 17240289.
Fuentes bibliográficas
- National genome institute
- Andrich, Jürgen y Jörg T. Epplen, "Enfermedad de Huntington", Mente y Cerebro, 17, 2006, págs. 78-82.
- Thompson, M.W., McInnes, R.R., Willard, H.F., "Thompson & Thompson. Genética Médica". 7ª edición, Editorial Elsevier Masson.
- Jorde, L.B., Carey,J.C., Bamshad, M.J., White,R.L., "Medical Genetics". 2ª edición, Editorial Harcourt.
- Solari, A.J., "Genética humana: fundamentos y aplicaciones en medicina". 3ª edición, Editorial Panamericana.
- OMIM. Huntington disease
Bibliografía sobre la enfermedad
- Rodes Huntington´s Disease: Hope Through Research. National Institute of Neurological Disorders and Stroke (NINDS).
- Degenerative (Huntington´s) Chorea. Clinical Neurology on CD-ROM. Lippincott-Raven. 1996
- Bird DE. Huntington´s chorea: etiology and pathogenesis. Handbook of clinical neurology. En: P.J.Vinken, G.W. Bruyn and H.L.Klawans, editors. Extrapyramidal disorders. Elsevier science publishers BV.1986;5(49):255-299
- Roos RA. Neuropathology of Huntington´s chorea. Handbook of clinical neurology. En: P.J.Vinken, G.W. Bruyn and H.L.Klawans, editors. Extrapyramidal disorders. Elsevier science publishers BV.1986;5(49):315-325
- Ross C, Margolis R, Rosenblatt A, et al. Huntington disease and related disorder, dentatorubral-pallidoluysian atrophy (DRPLA). Medicine. 1997;76(5):305-329
- Schapira AH. Mitochondrial function in Huntington´s disease: clues for pathogenesis and prospects for treatment. Ann Neurol. 1997;41(2):141-142
- Browne SE, Bowling AC, McGarvey A, et al. Oxidative damage and metabolic dysfunction in Huntington´s disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646-653
- Fernández-Álvarez E. Corea y atetosis en la infancia. Rev Neurol (Barc) 1995;23(supl 3):S 330-S 333
- Chutorian A. Distonías. Rev Neurol(Barc) 1995;23(supl 3):S 339-S 348
- Jiménez-Jiménez FJ, Ortí-Pareja M, Molina-Arjona JA. Alteraciones mitocondriales en las enfermedades neurodegenerativas. Rev Neurol 1998;26(supl 1):S 112-S 117
- Deus-Yela J, Pujol J, Espert R. Deterioro neuropsicológico en la enfermedad de Huntington. Rev Neurol 1997;25(144):1257-1268
- Burguera JA, Solís P, Salazar A. Estimación de la prevalencia de la enfermedad de Huntington por el método de captura-recaptura en la Comunidad Valenciana. Rev Neurol 1997;25(148):1845-1847
- Nance MA, and the US Huntington Disease Genetic Testing Groupe. Genetic testing of children at risk for Huntington´s Disease. Neurology 1997;49:1048-1053
- Snowden JS, Craufurd D, Griffiths HL, Neary D. Awareness of involuntary movements in Huntington Disease. Arch Neurol 1998;55:801-805
- Gómez-Tortosa E, del Barrio A, García PJ, et al. Severity of congnitive impairment in juvenile and late-onset Huntington disease. Arch Neurol 1998;55:835-843
Véase también
Enlaces externos
- http://www.nlm.nih.gov/medlineplus/ spanish/ency/article/000000.htm
- http://www.scn.es/cursos/tmovimiento/CAPITULO_X.htm
- Huntington's Disease Society of America en la dirección: http://hdsa.org/.
- www.discapnet.es/Discapnet/Castellano/ Salud/Enfermedades/C/Corea+de+Huntington/
- Sistema de Información sobre enfermedades raras en español, iier.isciii.es/er/prg/er_bus2.asp?cod_enf=1309
- http://e-huntington.org/. (Asociación Corea de Huntington Española - ACHE)
- http://es.hdbuzz.net - HDBuzz: Novedades en la investigación en la EH, en lenguaje sencillo, escrito por investigadores.
Categorías:- Enfermedades hereditarias
- Enfermedades neurológicas
- Enfermedades degenerativas
- Enfermedades raras
- Enfermedades epónimas
- Demencias



Wikimedia foundation. 2010.