- Síndrome de Charcot–Marie–Tooth
-
Síndrome de Charcot–Marie–Tooth 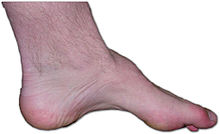
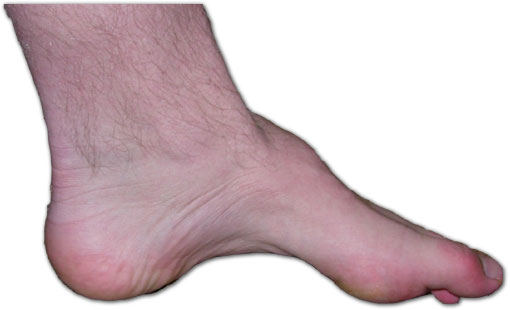
Pie de un paciente con la enfermedad Charcot-Marie-Tooth. La falta de músculo, el arco alto, y signo de martillo, son signos de esta enfermedad genéticaClasificación y recursos externos CIE-10 G 60.0; g 60 CIE-9 356.1 DiseasesDB 222 MedlinePlus Información de salud en la enciclopedia MedlinePlus PubMed Buscar en Medline mediante PubMed (en inglés) eMedicine ortopedia/43 eMedicine2 pmr 29 MeSH D002607  Aviso médico
Aviso médico La enfermedad de Charcot-Marie-Tooth comprende un grupo heterogéneo de neuropatías periféricas hereditarias no inflamatorias. También se le puede llamar como Atrofia muscular peroneal, neuropatía motora y sensorial hereditaria. Suele comenzar a los 10 ó 20 años, pero a veces lo hace más tarde, hasta los 50-60 años.
Contenido
Síntomas
La presentación varía según las distintas familias, pero los individuos afectados de una familia tienden a mostrar una sintomatología similar. Por lo general, comienzo gradual con progresión lenta, deformidad del pie que produce arco alto (cavo) y dedos en martillo, atrofia de las piernas que origina un aspecto de patas de cigüeña, agrandamiento de los nervios, pérdida sensorial u otros signos neurológicos, escoliosis, división de la propiocepción, que muchas veces interfiere en el equilibrio y la marcha, parestesis dolorosas, (en casos avanzados), posible afectación de las manos, ausencia de reflejos tendinosos profundos en muchos pacientes, úlceras de los pies,la cual puede tardar 1 año en volver a regenerarse,debido al debil tejido de la planta del pie, con escasa tendencia a la cicatrización en algunos casos.
Etiología
Desmielización segmentaria crónica de los nervios periféricos, con cambios hipertróficos causados por la remielinación.
Diagnóstico
Diagnóstico diferencial: otras neuropatías hereditarias. Polineuropatías tóxicas, metabólicas y nutricionales.
Valoración
El comienzo temprano, la progresión lenta y la naturaleza familiar del trastorno suelen ser suficientes para establecer el diagnóstico. Los estudios electrofisiológicos acostumbran a ser diagnósticos y también pueden ser útiles para definir varios subtipos de este grupo de neuropatías. En ocasiones, se requieren biopsias de músculo y nervio (sural).
Tratamiento
- A corto plazo: consejo genético. Fisioterapia de apoyo y terapia laboral. Prevención de las lesiones en los miembros con sensibilidad disminuida. Empleo de ortesis.
- A largo plazo: la cirugía mejora a veces la estabilidad y restaura un pie plantígrado.
Evolución y Pronóstico
La incapacidad suele ser leve y compatible con una vida larga. El 10-20% de los pacientes permanecen asintomáticos. Un pequeño número de pacientes pierden la capacidad para caminar en la sexta o la séptima décadas de la vida.
Derivación
Consulta ortopédica para la aplicación de ortesis y tratamiento de la deformidad. Consejo genético.
Categorías:- Enfermedades hereditarias
- Enfermedades neurológicas
- Enfermedades epónimas
Wikimedia foundation. 2010.