- Síndrome de Klinefelter
-
Síndrome de Klinefelter 
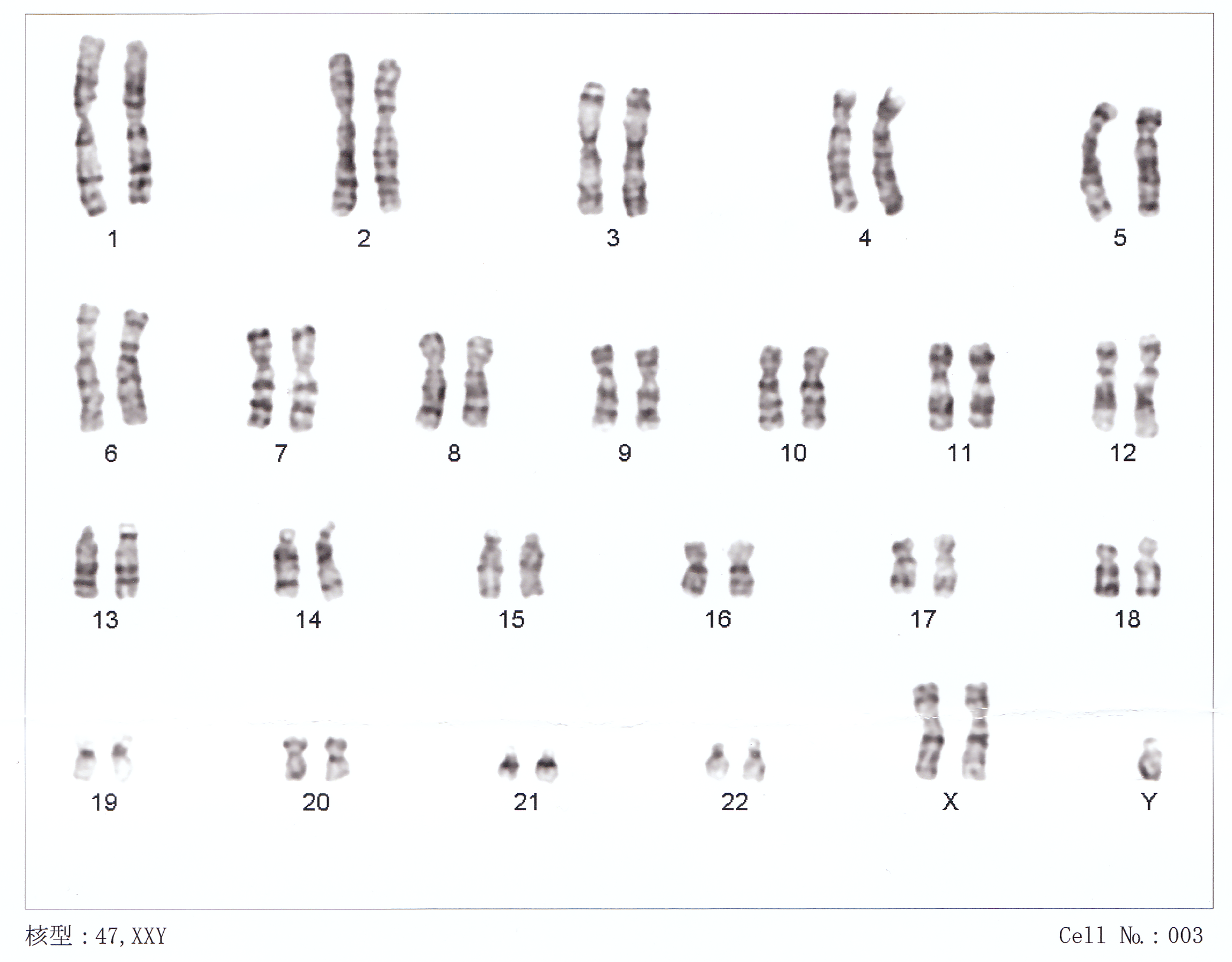
47,XXYClasificación y recursos externos CIE-10 Q98.0-Q98.4 CIE-9 758.7 eMedicine ped/1252 MeSH D007713  Aviso médico
Aviso médicoEl síndrome de Klinefelter es una anomalía cromosómica que afecta solamente a los hombres y ocasiona hipogonadismo.
El sexo de las personas está determinado por los cromosomas X y Y. Los hombres tienen los cromosomas 44XY (46) y las mujeres tienen los cromosomas 44XX (46). En el síndrome de Klinefelter se pueden presentar los cromosomas 44XXY (47), 44XXXY (48), 44XXYY(48), 44XXXXY (49), etc y los llamados "mosaicos" o "mosaicismos" 46XY / 47XXY.
Es una alteración genética que se desarrolla por la separación incorrecta de los cromosomas homólogos durante las meiosis que dan lugar a los gametos de uno de los progenitores, aunque también puede darse en las primeras divisiones del cigoto.
Se cree que Carlos II de España sufrió este síndrome, debido fundamentalmente a los sucesivos matrimonios endogámicos de sus antepasados y falta de sangre nueva.
Contenido
Genética
El síndrome de Klinefelter o disgenesia de los túbulos seminíferos se considera la anomalía cromosómica más común en los humanos, presentándose con una incidencia de 1 en 500 en los recién nacidos vivos varones. Los afectados presentan un cromosoma “X” supernumerario lo que conduce a fallo testicular primario con infertilidad e hipoandrogenismo. A pesar de la relativa frecuencia del padecimiento en recién nacidos vivos, se estima que la mitad de los productos 47, XXY se abortan de manera espontánea.
El síndrome de Klinefelter es considerado la causa más frecuente de hipogonadismo hipergonadotrópico.
Fue descrito en 1942 por Klinefelter y colaboradores que estudiaron 9 pacientes con: ginecomastia, testículos pequeños, azoospermia, cifras elevadas de gonadotropinas. Ellos sugirieron que el defecto primario estaba en las células de Sertoli y propusieron que además en estos pacientes había una deficiencia en una hormona testicular que regulaba la concentración de gonadotropinas hipofisiarias, a la que llamaron hormona “X” o inhibina.
En 1956 se demostró la presencia del corpúsculo de Barr en pacientes con síndrome de Klinefelter y en 1959 se identifica que el cariotipo de un sujeto con la enfermedad era 47, XXY. De esta manera se estableció que la presencia de un cromosoma “X” extra era el factor etiológico fundamental.
Manifestaciones
No todas estas manifestaciones se dan en un mismo individuo:
- Talla elevada
- Mayor acumulación de grasa subcutánea
- Dismorfia facial discreta
- Alteraciones dentarias
- En ocasiones criptorquidia, micropene, escroto hipoplásico o malformaciones en los genitales.
- Esterilidad por azoospermia.
- Ginecomastia uni o bilateral
- Vello pubiano disminuido
- Gonadotrofinas elevadas en la pubertad
- Disminución de la libido
- Retraso en el área del lenguaje, lectura y comprensión
- Lentitud, apatía.
- Trastornos emocionales, ansiedad, depresión, etc.
- Falta de autoestima.
Patogénesis
El cromosoma adicional en los pacientes con síndrome de Klinefelter a menudo es adquirido por un error de disyunción durante la gametogénesis de alguno de los padres, originando gametos con 24 cromosomas debido a un cromosoma “X” supernumerario.
Un 56% de individuos 47, XXY se debe a no disyunción materna (el cromosoma “X” supernumerario proviene de la madre), el 44% son debidos a errores en la meiosis paterna como ocurren las trisomías autosómicas; la no disyunción se relaciona con edad materna avanzada. La anomalía cromosómica puede originarse también por un error durante las divisiones mitóticas del cigoto, produciendo así los casos de mosaicismo.
Los estudios en sujetos prepuberales 47, XXY no muestran deficiencias en las concentraciones de LH, FSH o testosterona, comparados con sujetos prepúberes 46, XY y la respuesta a la gonadoliberina (LHRH, hormona hipotalámica liberadora de gonadotropinas) es normal en ambos grupos. Sin embargo entre los 12 y 14 años de edad en los sujetos 47 XXY, las concentraciones de gonadotropinas se incrementan y la testosterona permanece en límites inferiores para la edad.
En biopsias realizadas a niños con el síndrome se ha observado sólo disminución en el número de células germinales. No obstante después de la pubertad se aprecia hialinización y fibrosis de los túbulos seminíferos, que son los cambios histológicos característicos del síndrome y que originan disminución en el volumen testicular y aumento de su consistencia. Además se observa ausencia de células germinales e hiperplasia y agregación de las células de Leydig, como repuesta a hiperestimulación por la LH. Las alteraciones histológicas se hacen más frecuentes con la edad.
La pérdida de túbulos seminíferos y células de Sertoli produce una disminución en las cifras de inhibina B, el factor regulador de FSH, y de AMH u hormona antimülleriana, lo que disminuye la retroalimentación negativa sobre la FSH, aumentando esta. La ausencia de espermatogénesis es secundaria a la presencia de cromosomas supernumerarios que se mantienen activos durante la gametogénesis.
Véase también
Enlaces externos
Categorías:- Síndromes
- Enfermedades genéticas
- Enfermedades masculinas
- Enfermedades del aparato reproductor masculino
- Enfermedades epónimas
Wikimedia foundation. 2010.