- Síndrome de Usher
-
Síndrome de Usher Clasificación y recursos externos OMIM 276900 DiseasesDB 13611 MeSH D052245 Sinónimos Síndrome de Hallgren
Síndrome de Usher-Hallgren Aviso médico
Aviso médico El síndrome de Usher (también conocido como síndrome de Hallgren o síndrome de Usher-Hallgren) es un raro trastorno genético asociado a una mutación en uno de los diez genes determinantes vinculados al mismo. Es la principal causa de sordoceguera congénita y se hereda según un patrón autosómico recesivo.[1]
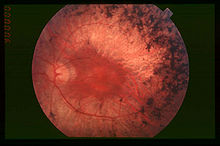
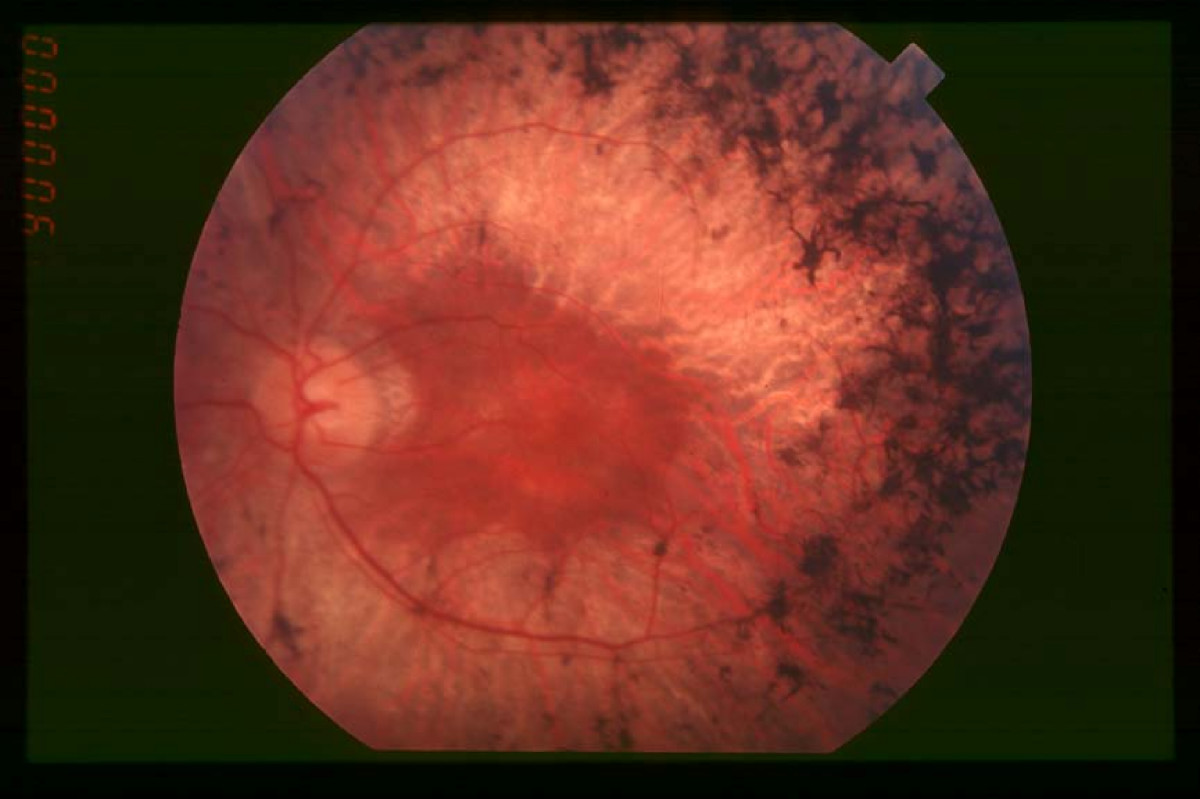
Este síndrome se caracteriza por sordera y pérdida gradual de la vista. Los problemas auditivos se deben a un defecto en el oído interno, que en ocasiones puede afectar también al sistema vestibular, mientras que la deficiencia visual se asocia con retinosis pigmentaria (RP), una degeneración de las células de la retina.[2]
Contenido
Historia
El nombre del síndrome se dio en honor del oftalmólogo escocés Charles Usher, quien examinó en el año 1914 numerosos pacientes afectos de esta patología y determinó su carácter hereditario.[3] [4] Sin embargo, fue descrito por primera vez en 1858 por Albrecht von Gräfe, un pionero de la oftalmología moderna, en un caso de un paciente sordo con retinosis pigmentaria, que tenía dos hermanos con los mismos síntomas.[5]
Tres años más tarde, uno de sus alumnos, Richard Liebreich, examinó la población judía de Berlín con sordera asociada a retinosis pigmentaria, y observó que la mayoría de los individuos afectados se trataban de hijos de matrimonios consanguíneos o pertenecientes a familias con antecedentes de la misma condición.[6] De estos hallazgos se podía deducir una herencia combinada de sordera y ceguera.
Epidemiología
El síndrome de Usher es responsable de la mayoría de los casos de sordoceguera congénita.[7] La prevalencia global estimada es de 3,5 afectados por cada 100.000 personas,[8] aunque ésta varía dependiendo de los países o regiones concretas en los que existen estudios epidemiológicos.
En España, la prevalencia se estima en 4,2 por cada 100.000 niños nacidos vivos,[9] mientras que en Colombia es de 3,2 por cada 100.000 habitantes, donde el 9% de los sordos padecen el síndrome.[10] En los Estados Unidos, se presenta en aproximadamente 1 persona de cada 23.000,[11] 1 de cada 28.000 en Noruega[12] y 1 de cada 12.500 en Alemania.[13] En Venezuela actualmente están haciéndose estudios epidemiológicos, destacando la altísima prevalencia de síndrome de Usher en la península de Macanao, que, con 76 afectados por cada 100.000 habitantes, es la mayor de América Latina.[14]
Los afectados con síndrome de Usher representan aproximadamente una sexta parte de las personas con retinosis pigmentaria. El Usher I y II son las formas más comunes; la fracción de las personas con Usher III es significativa sólo en unas pocas áreas específicas, tales como Finlandia[15] y Birmingham.[16] La forma predominante en España es la del tipo II, con un 65% de los afectados, siendo el tipo I el restante 35%.[17] Un estudio de los pacientes colombianos con Usher reveló que el tipo I es el más común, con alrededor del 70% de los afectados, teniendo una menor incidencia el tipo II (26%) y el III (4%).[18]
Clasificación y etiopatogenia
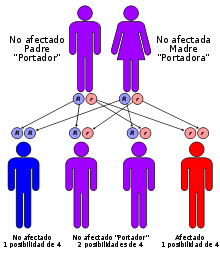 Herencia autosómica recesiva.
Herencia autosómica recesiva.
Su clasificación se basa en el grado de sordera, edad a la que aparece la ceguera y la posible afectación al sistema vestibular,[9] pero también a su vez, estos subtipos clínicos pueden subdividirse por el gen mutado en particular; las personas con Usher I tienen seis genes determinantes y una mutación en cualquiera de ellos produce la enfermedad. En el tipo II están implicados tres genes, pero basta con la mutación de uno solo de ellos, mientras que sólo un gen se ha asociado con el Usher III.
Como el síndrome de Usher se hereda de manera autosómica recesiva,[9] tanto hombres como mujeres tienen las mismas probabilidades de heredar este síndrome, teniendo los hijos de padres que ambos son portadores de la misma mutación un cuarto de posibilidades de heredar la enfermedad y los niños de esos padres que no resultan afectados tienen una probabilidad de dos tercios de ser portadores. Así mismo, los hijos de padres que sólo uno de ellos es portador no tienen probabilidad de tener la enfermedad, pero sí de ser portador. Por lo tanto, la consanguinidad de los padres es un factor de riesgo.
Por lo general, los bastones de la retina se ven afectados en primer lugar, lo que conduce a ceguera nocturna (nictalopía) y a una pérdida gradual de visión periférica. En otros casos, hay degeneración temprana de los conos en la mácula, lo que lleva a una pérdida de agudeza visual central. En algunos casos, la visión de la fóvea está a salvo dando lugar a la visión de "donut" o en túnel del paciente: la visión central está intacta, pero hay un anillo alrededor de ella en el que la visión se deteriora.
El síndrome de Usher I
Las personas con Usher I normalmente nacen sordas y con frecuencia tienen dificultades para mantener su equilibrio debido a problemas en el sistema vestibular. Los bebés con Usher I suelen ser lentos para desarrollar habilidades motoras tales como caminar. Además, tienen dificultades de hacerse entender con el habla. Los problemas de visión comienzan a desarrollarse antes de la pubertad.[9]
El síndrome de Usher tipo I puede ser causado por mutaciones en uno de varios genes determinantes diferentes: CDH23,[19] MYO7A,[20] PCDH15,[21] USH1C[22] y USH1G.[23] Estos genes funcionan en el desarrollo y mantenimiento del oído interno, en estructuras tales como los estereocilios, que transmiten el sonido y el movimiento en señales al cerebro. Las alteraciones en estos genes pueden causar una incapacidad para mantener el equilibrio (disfunción vestibular) y la pérdida de audición. Estos genes también juegan un papel en el desarrollo y mantenimiento de la retina al influir en la estructura y función de las células fotorreceptoras y en células de soporte del llamado epitelio pigmentado de la retina. Las mutaciones que afectan la función normal de estos genes pueden dar lugar a retinosis pigmentaria y a pérdida de la visión que comienza a darse en la primera década de vida. El tipo I es más común en personas de ascendencia judía asquenazí y en la población francesa de Acadia (Luisiana).[24] Otro gen en el cromosoma 21 se haya implicado en el Usher tipo I, identificado con el símbolo USH1E pero su función aún es desconocida. Ésta última variedad se ha descrito tan solo en una familia marroquí.[25]
El síndrome de Usher II
Los afectados con Usher II son generalmente duros de oído en vez de sordos, su capacidad auditiva no se degrada con el tiempo y usualmente tienen un sistema vestibular normal. El síndrome de Usher tipo II se da al menos con tanta frecuencia como el tipo I, sin embargo, el tipo II podría ser bastante más común que el tipo I, pero suele ser mal diagnosticado debido a la mayor dificultad de describir los síntomas. La pérdida de la visión generalmente se da en la segunda década de vida.[9]
El síndrome de Usher tipo II puede ser causado por mutaciones en cualquiera de los tres genes diferentes implicados: USH2A, GPR98 y DFNB31. La proteína codificada por el gen USH2A, la usherina, se expresa en la membrana basal de la cóclea y la retina, entre otros tejidos. La usherina es fundamental para el correcto desarrollo y mantenimiento de estas estructuras, lo que podría explicar su papel en la pérdida de audición y de visión.[26] El segundo gen codifica un receptor transmembrana localizado en el sistema nervioso central,[27] mientras que el tercero codifica la whirlina, una proteína que parece implicada en la estabilidad y mantenimiento de las proyecciones de los estereocilios del órgano de Corti y en la formación del citoesqueleto de actina de éstas células.[28]
 Comparación Gravedad
Comparación Gravedad
Retinosis Pigmentaria[29]Usher I Usher II Ceguera Nocturna 9.2 ± 7.3 años 17.2 ± 8.7 años Constricción Campo Visual 10.2 ± 6.6 años 20.2 ± 10.2 años Disminución Agudeza Visual 16.9 ± 12.7 años 24.9 ± 14.9 años El síndrome de Usher III
Por el contrario, las personas con Usher tipo III padecen una progresiva pérdida de la audición y aproximadamente la mitad tiene una disfunción vestibular. La pérdida de la visión también es progresiva.[9] La frecuencia del síndrome de Usher tipo III es el más alto en la población finlandesa, pero se ha observado raramente en otros grupos étnicos, como en judíos de ascendencia asquenazí.[30] [31]
Las mutaciones en un solo gen, el CLRN1, se han relacionado con el síndrome de Usher tipo III. El gen codifica la clarina-1, una proteína que parece ser importante para el desarrollo y la homeostasis del oído interno y la retina.[32] Además, podría jugar un papel esencial en la excitación durante la sinapsis en la percepción del sonido, visión y equilibrio.[33]
Diagnóstico
Como el síndrome de Usher es incurable en la actualidad, es útil para los niños diagnosticarlo mucho antes de que desarrollen la ceguera nocturna característica. Algunos estudios preliminares han sugerido que hasta el 10% de los niños sordos congénitos pueden padecer ésta enfermedad.[1] El síndrome de Usher puede sospecharse si el niño es sordo profundo desde el nacimiento y lento, sobre todo para caminar. La consanguinidad de los padres es un factor importante a tener en cuenta en el historial. Un diagnóstico precoz es fundamental para cubrir las necesidades pedagógicas especiales que pueda tener un afectado de sordoceguera y ofrecer una orientación informativa acerca de la situación en la que se encuentra, además del apoyo psicológico que precisen tanto el paciente como sus familiares.
En el diagnóstico del síndrome se incluyen diferentes pruebas que valoran las distintas características de la enfermedad. Así, para la sordera se utiliza un audímetro u otras pruebas de audición que evalúan la sensibilidad auditiva del paciente a diferentes frecuencias.[34] La electronistagmografía (ENG) se utiliza para detectar los posibles problemas de equilibrio. Ésta técnica consiste en colocar electrodos alrededor de los ojos, además de otro en la frente, que registran los movimientos oculares (nistagmos) cuando se estimula el oído interno y los nervios circundantes al verter agua caliente (o tibia) y fría en el conducto auditivo externo en diferentes momentos. Usando el agua fría, los ojos deben moverse rápido hacia el lado opuesto al oído en el que se ha instilado el líquido, retornando lentamente. Con el agua caliente ocurre al contrario, se acercan rápidamente para luego alejarse despacio. Cada oído se examina individualmente. Los resultados anormales indican daños en el oído interno, en el nervio auditivo o el oculomotor, o en otras partes del cerebro.[35]
Un oftalmólogo se encarga de evaluar la visión del paciente mediante diferentes pruebas que incluyen la evaluación de la visión lateral, la habilidad de ver en oscuridad y la sensibilidad al color y al contraste. En caso de detectar algún problema, se utiliza el electrorretinograma (ERG), que comprueba, a través de un electrodo situado en cada ojo, la electricidad emitida por los nervios de la retina cuando el sujeto mira a una luz destellante. La prueba se realiza en condiciones de luz ambiental y de oscuridad, y sirve para identificar un caso de retinitis pigmentaria.[34]
Para confirmar totalmente si se trata del síndrome de Usher, se puede recurrir a un estudio genético que desvele las características mutaciones cromosómicas. Hasta el momento, se han detectado más de 400 mutaciones diferentes en los genes implicados en la enfermedad.[36] Para llevar a cabo el análisis de las mutaciones, se extrae ADN de una muestra de sangre del paciente. De este ADN se seleccionan los exones en los que se han detectado mutaciones que producen síndrome de Usher y a través de la técnica de PCR se copian estos fragmentos hasta lograr un tamaño adecuado para el análisis de su secuencia en un laboratorio. Éstas secuencias se comparan con las secuencias patrón conocidas.[37] [38]
Diagnóstico diferencial
Otros trece síndromes pueden presentar síntomas similares al síndrome de Usher:
- El síndrome de Alport: se caracteriza por el daño de los glomérulos renales, apareciendo hematuria y proteinuria, detectables con un análisis de orina.[39]
- El síndrome de Alström: para descartar esta enfermedad es necesario un análisis de la glucemia, ya que la sordoceguera está acompañada de diabetes tipo 2, además de otras deficiencias metábolicas y una miocardiopatía.[40]
- El síndrome de Bardet-Biedl: en el diagnóstico de esta enfermedad multisistémica se deben describir varios de los signos primarios entre los que se encuentran anomalías renales, polidactilia, obesidad, dificultades de aprendizaje e hipogonadismo en varones.[41]
- El síndrome de Cockayne: los afectados por esta condición desarrollan durante las primeras etapas de su vida problemas en el crecimiento que conllevan una corta estatura y un peso excesivamente bajo, para terminar padeciendo alteraciones neurológicas.[42]
- la displasia congénita espondiloepifisaria: se puede distinguir por la corta estatura desde el momento del nacimiento, además de otros defectos esqueléticos que se desarrollan durante la infancia.[43]
- El síndrome de Flynn-Aird: desde edades tempranas se puede diagnosticar esta enfermedad, en el que el paciente sufre alteraciones óseas (cifosis, cifoescoliosis), musculares (hipotrofia), neuronales (afasia, pérdida de reflejos, convulsiones) y atrofia dérmica o subcutánea; las alteraciones visuales son debidas mayoritariamente a una miopía severa o a cataratas bilaterales, y en menor medida a retinitis pigmentosa.[44]
- La ataxia de Friedreich: afecta más a la médula espinal y raíces espinales, provocando alteraciones motores, pérdida de los reflejos tendinosos y de sensibilidad, mientras que la sordoceguera es ocasional.[45]
- El síndrome de Hurler (MPS-1): destacan como síntomas principales en el momento del diagnóstico el retraso mental, complicaciones respiratorias y ortopédicas.[46]
- El síndrome de Kearns-Sayre (CPEO): los niños afectados suelen desarrollar endocrinopatías como diabetes, hipoparatiroidismo, la enfermedad de Adisson y alteraciones en el crecimiento; otros signos incluyen alteraciones musculares, cardíacas y en el sistema nervioso central.[47]
- La enfermedad de Norrie: la ceguera producida por esta enfermedad es debida al crecimiento de una masa tras la pupila, siendo la leucocoria el primer signo visible.[48]
- La osteopetrosis (enfermedad de Albers-Schonberg): los síntomas mayoritarios de esta enfermedad son óseos y hematopoyéticos debido a un aumento de la densidad de los huesos, visible a través de radiografías.[49]
- La enfermedad de Refsum (enfermedad por almacenamiento de ácido fitánico): este desorden es debido a una deficiencia en la enzima que degrada el ácido fitánico, por lo que se puede diagnosticar mediante un análisis de sangre que detecte niveles altos de ese ácido graso.[50]
- El síndrome de Zellweger (síndrome cerebro-hepato-renal): aparte de la sordoceguera, en el diagnóstico clínico se observan rasgos faciales anormales, hepatomegalia, niveles elevados de aluminio y hierro en sangre y retraso mental.[51]
Tratamiento
Como se ha dicho anteriormente, hoy en día el síndrome de Usher no tiene remedio, aunque se pueden usar diferentes terapias para mejorar la vida de los pacientes tratando la falta de audición y de visión por separado. Estos tratamientos tienen mejores resultados en niños que en adultos. De este modo, para tratar la sordera, se coloca un implante coclear en el oído interno mediante cirugía, que transforma los sonidos en señales eléctricas estimulando en nervio auditivo. Éste tratamiento ha dado buenos resultados en niños menores de 3 años. En el caso de la retinosis pigmentaria, el tratamiento con vitamina A en algunos pacientes ha conseguido ralentizar la progresión de la enfermedad.[52]
Dado que el síndrome de Usher es resultado de la pérdida de un gen, la terapia génica que añade el gen adecuado puede aliviarlo, siempre que las proteínas sintetizadas funcionen. Estudios recientes en ratones de laboratorio han mostrado que una forma de la enfermedad que se asocia con la mutación en MYO7A, puede atenuarse reemplazando el gen mutante con un lentivirus.[53] Sin embargo, algunos de los genes mutados asociados con el síndrome de Usher codifican proteínas muy grandes, como las proteínas de los genes USH2A y GPR98, que tienen aproximadamente 6.000 aminoácidos,[54] [55] por lo que puede ser complicado obtener buenos resultados con esta terapia.
Existen otras vías alternativas que actualmente se encuentran en desarrollo y podrían ser un tratamiento eficaz aplicable al síndrome de Usher. La tecnología de células encapsuladas se encuentra en fase de pruebas, pudiendo estar disponible en esta década. Esta técnica consiste en un implante intraocular, una microcápsula de unos 10 milímetros, que contiene células humanas modificadas genéticamente para secretar el llamado factor neurotrófico ciliar (CNTF), una citoquina que estimula la expresión génica, diferenciación y regeneración de las neuronas sensitivas y motoras, y que ha demostrado ser eficaz en el retraso de la degeneración neuronal en diferentes enfermedades.[56] Éste es liberado directamente en la parte posterior del ojo de una forma controlada y continua, eludiendo así la barrera hemato-retiniana y superando uno de los obstáculos principales en el tratamiento de las enfermedades de la retina.[57]
Por último, la terapia con células madre, que se encuentra en fase experimental en un gran número de campos, ha sido utilizada en el tratamiento de la sordera y la ceguera.[58] [59] En un futuro próximo, el uso de una de éstas técnicas o una combinación de todas, podría ser la solución a este síndrome.[52]
Referencias
- ↑ a b Mets MB, Young NM, Pass A, Lasky JB (2000). «Early diagnosis of Usher syndrome in children». Transactions of the American Ophthalmological Society 98: pp. 237–45. PMID 11190026.
- ↑ Fishman GA, Kumar A, Joseph ME, Torok N, and Andersonj RJ (1983). «Usher's syndrome». Archives of Ophthalmology 109 (9): pp. 1367–1374. PMID 6604514.
- ↑ Usher, Charles (1914). «On the inheritance of Retinitis pigmentosa with notes of cases.». Roy.Lond.Ophthalmol.Hosp.Rep. 19: pp. 130-236.
- ↑ Usher, Charles (1935). «Bowman lecture: On a few hereditary eye affection.». Trans Ophthlamol Soc UK 55: pp. 164.
- ↑ von Gräfe A (1858). «Exceptionelles Verhalten des Gesichtsfeldes bei Pigmententartung der Netzhaut». Archiv für Ophthalmologie 4: pp. 250–253.
- ↑ Liebreich R (1861). «Abkunft aus Ehen unter Blutsverwandten als Grund von Retinitis pigmentosa». Dtsch. Klin. 13: pp. 53.
- ↑ Vernon M (1969). «Usher,s syndrome — deafness and progressive blindness. Clinical cases, prevention, theory and literature survey». Journal of Chronic Disorders 22 (3): pp. 133–151. doi:. PMID 4897966.
- ↑ Prevalence of rare diseases: Bibliografic data, Informes periódicos de Orphanet, Serie de Enfermedades raras, Noviembre de 2010, Nº 1: Lista por orden alfabético
- ↑ a b c d e f Millán, José M. «El Síndrome de Usher: diagnóstico de una enfermedad geneticamente hetereogénea y la importancia del estudio poblacional». Unidad de genética del Hospital de la Fe, Valencia. CIBERER. http://www.segenetica.es/curso_g_humana/MILLAN_JOSEM.PDF.
- ↑ Tamayo Fernández, Mª Lucía (1999). Análisis Etiológico, Médico – Genético, Estadístico y epidemiológico de la Limitación Visual en Colombia.
- ↑ Boughman J, Vernon M, Shaver K (1983). «Usher syndrome: Definition and estimate of prevalence from two high-risk populations». Journal of Chronic Disorders 36 (8): pp. 595–603. doi:. PMID 6885960.
- ↑ Grøndahl J (1987). «Estimation of prognosis and prevalence of retinitis pigmentosa and Usher syndrome in Norway». Clin. Genet. 31 (4): pp. 255–264. doi:. PMID 3594933.
- ↑ Otterstedde CR, Spandau U, Blankenagel A, Kimberling WJ, Reisser C (2001). «A new clinical classication for Usher,s syndrome based on a new subtype of Usher,s syndrome type I». Laryngoscope 111 (1): pp. 84–86. doi:. PMID 11192904.
- ↑ SOCIEVEN (ed.): «Laboratorio de Génetica Sobre el Síndrome de Usher» (en español). Consultado el 23 de diciembre de 2010.
- ↑ Pakarinen L, Tuppurainen K, Laipapala P, Mäntyjärvi M, Puhakka H (1996). «The ophthalmological course of Usher syndrome type III». International Ophthalmology 19 (5): pp. 307–311. doi:. PMID 8864816.
- ↑ Hope CI, Bundey S, Proops D, Fielder AR (1997). «Usher syndrome in the city of Birmingham — prevalence and clinical classification». British Journal of Ophthalmology 81 (1): pp. 46–53. doi:. PMID 9135408.
- ↑ Millán (24 febrero del 2007). «Detectan un nuevo gen implicado en el síndrome de Usher.» (en español). Consultado el 23 de diciembre de 2010.
- ↑ Tamayo ML, Bernal JE, Tamayo GE, Frías JL, Alvira G, Vergara O, Rodríguez V, Uribe JI, Silva JC (1991). «Usher syndrome: results of a screening program in Colombia.». Clinical genetics 40 (4). PMID 1415347.
- ↑ Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CR, Wayne S, Bellman S, Desmukh D, Ahmed Z, Khan SN, Kaloustian VM, Li XC, Lalwani A, Riazuddin S, Bitner-Glindzicz M, Nance WE, Liu XZ, Wistow G, Smith RJ, Griffith AJ, Wilcox ER, Friedman TB, Morell RJ (enero de 2001). «Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23». American journal of human genetics 68: pp. 26–37. doi:. PMID 11090341.
- ↑ Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD (1995). «Defective myosin VIIA gene responsible for Usher syndrome type 1B». Nature 374 (6517): pp. 60–1. doi:. PMID 7870171.
- ↑ Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, Morell RJ, Friedman TB, Riazuddin S, Wilcox ER (2001). «Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F». American journal of human genetics 69 (1): pp. 25–34. doi:. PMID 11398101.
- ↑ Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJ, Slim R, Petit C (octubre de 2000). «A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C». Nature Genetics 26 (1): pp. 51–5. doi:. PMID 10973247.
- ↑ Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Laine S, Delmaghani S, Adato A, Nadifi S, Zina ZB, Hamel C, Gal A, Ayadi H, Yonekawa H, Petit C (Feb 2003). «Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin». Human molecular genetics 12 (5): pp. 463–71. doi:. PMID 12588794.
- ↑ Smith RJ, Pelias MZ, Daiger SP, Keats B, Kimberling W, Hejtmancik JF (1992). «Clinical variability and genetic heterogeneity within the Acadian Usher population.». American journal of medical genetics 43 (6). PMID 1415347.
- ↑ Chaïb H, Kaplan J, Gerber S, Vincent C, Ayadi H, Slim R, Munnich A, Weissenbach J, Petit C (1997). «A newly identified locus for Usher syndrome type I, USH1E, maps to chromosome 21q21». Human molecular genetics 6 (1). PMID 9002666.
- ↑ Eudy J.D., Weston M.D., Yao S.F., Hoover D.M., Rehm H.L., Ahmad I., Ma-Edmonds M., Yan D., Cheng J.J., Beisel K.W., Ayuso C., Cremers C., Davenport S., Moller C., Talmadge C.B., Tamayo M., Swaroop A., Morton C.C., Kimberling W.J., Sumegi J. (1998). «Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa». Science 280 (5370). PMID 9624053.
- ↑ «G-protein coupled receptor 98 - Homo sapiens». UniProt Knowledgebase. Consultado el 9 de enero de 2011.
- ↑ «Whirlin - Homo sapiens». UniProt Knowledgebase. Consultado el 9 de enero de 2011.
- ↑ Ayuso, Carmen; García-Sandoval, Blanca; Gutiérrez, Raimundo; Cenjor, Carlos. «Síndrome de Usher: Estudio clínico y genético en España». Fundación Jiménez Díaz, Madrid.
- ↑ Joensuu T, Hämäläinen R, Yuan B, Johnson C, Tegelberg S, Gasparini P, Zelante L, Pirvola U, Pakarinen L, Lehesjoki AE, de la Chapelle A, Sankila EM (2001). «Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3». American journal of human genetics 69 (1160). PMID 11524702.
- ↑ Fields RR, Zhou G, Huang D, Davis JR, Möller C, Jacobson SG, Kimberling WJ, Sumegi J. (2002). «Usher syndrome type III: revised genomic structure of the USH3 gene and identification of novel mutations». American journal of human genetics 71. PMID 12145752.
- ↑ Pruitt KD, Tatusova T, Maglott DR. «Clarin-1 - Homo sapiens» (en inglés). The Reference Sequence (RefSeq) Project. Consultado el 8 de enero de 2011.
- ↑ Fields RR, Zhou G, Huang D, Davis JR, Moeller C, Jacobson SG, Kimberling WJ, Sumegi J (2002). «Usher syndrome type III: revised genomic structure of the USH3 gene and identification of novel mutations». American journal of human genetics 71. PMID 12145752.
- ↑ a b Laurie Rosenblum, MPH (12 de enero de 2010). «Síndrome de Usher» (en español). Consultado el 24 de diciembre de 2010.
- ↑ Griggs RC, Jozefowicz RF, Aminoff MJ. (2007). Cecil medicine (23ª edición). Saunders Elsevier.. http://www.nlm.nih.gov/medlineplus/spanish/ency/article/003448.htm. Consultado el 24 de diciembre de 2010. «Approach to the patient with neurologic disease.»
- ↑ «Investigadores de la Fe y del CIBERER han detecado 32 mutaciones causantes del síndrome de Usher en la población española» (en español) (Nota de prensa). Unidad de genética del Hospital de la Fe, Valencia. CIBERER. (4 de marzo de 2010). Consultado el 27 de enero de 2011.
- ↑ Guzmán H, Díaz de Palacios AM, Garrido E, Utrera R (2008). «Diagnóstico molecular del síndrome de Usher en Venezuela». Acta Otorrinolaringológica 20 (1). ISSN 0798-166X. http://www.svorl.org.ve/docs/Rev.ORL-nro1-2008.pdf.
- ↑ Vallcorba Gómez del Valle, I. «Diagnóstico molecular en genética médica». Ciencia, tecnología y medicina. http://www.jano.es/ficheros/sumarios/1/60/1384/57/1v60n1384a13012839pdf001.pdf.
- ↑ Saxena, R. (9 de septiembre de 2008). «Alport Syndrome: Differential Diagnoses & Workup» (en inglés). Consultado el 3 de enero de 2011.
- ↑ Kaneshiro, N.K. (27 de julio de 2010). «Síndrome de Alström» (en español). Medline plus. Consultado el 3 de enero de 2011.
- ↑ Izquierdo, M.; Avellaneda A. (febrero de 2004). «Bardet Biedl, Síndrome de» (en español). Consultado el 31 de diciembre de 2010.
- ↑ Flannery, David (24 de noviembre de 2009). «Cockayne Syndrome». Consultado el 31 de diciembre de 2010.
- ↑ Lyons Jones, Kenneth (2007) (en español). Características reconocibles de la malformacion humana (6ª edición). Elsevier. pp. 407. ISBN 978-84-8174-947-8. http://books.google.es/books?id=wWVVPThnScEC&pg=PA407&lpg=PA407&dq=displasia+espondiloepifisaria+cong%C3%A9nita&source=bl&ots=iAxZ-cps1O&sig=bALTc0uiJD4SQNSW1RHOrazMML4&hl=es&ei=TAwdTYLhFM618QPotaTiBQ&sa=X&oi=book_result&ct=result&resnum=1&ved=0CBYQ6AEwADgK#v=onepage&q=displasia%20espondiloepifisaria%20cong%C3%A9nita&f=false. Consultado el 30 de diciembre de 2010.
- ↑ Tamayo Fernadez, ML.; Bernal Villegas, J. (1998). «5» (en español). Alteraciones visuales y auditivas de origen genético: Aspectos oftalmológicos, audiológicos y genéticos. CEJA. pp. 152. ISBN 958-683-036-5. http://books.google.es/books?id=4LvAZ671N3MC&printsec=frontcover&dq=Alteraciones+visuales+y+auditivas+de+origen+gen%C3%A9tico&hl=es&ei=38AhTY2jBdWW4Ab9raiGAg&sa=X&oi=book_result&ct=result&resnum=1&ved=0CCkQ6AEwAA#v=onepage&q&f=false. Consultado el 3 de enero de 2011.
- ↑ Chawla, Jasvinder (18 de agosto de 2010). «Friedreich Ataxia». Consultado el 31 de diciembre de 2010.
- ↑ Correa Garzón, L.N.. «Mucopolisacarosis» (en español). Consultado el 31 de diciembre de 2010.
- ↑ Purna Basu, A.; Posner, E.; McFarland, R. (4 de febrero de 2010). «Kearns-Sayre Syndrome» (en inglés). Consultado el 1 de enero de 2011.
- ↑ Sims , Katherine. "NDP-Related Retinopathies." Gene Reviews. 8-8-2006.
- ↑ Bhargava, A.; Blank, R. (13 de octubre de 2009). «Osteopetrosis» (en inglés). Consultado el 1 de enero de 2011.
- ↑ Wright, Kenneth W.; Spiegel, Peter H. (2001) (en español). Pediatría y estrabismo. Harcourt. pp. 120. ISBN 84-8174-516-2. http://books.google.com/books?id=zFk8ND5WPBgC&pg=PA120&lpg=PA120&dq=refsum+enfermedad+enzima&source=bl&ots=WZljDTYSSj&sig=SV-khR3hy4m2oDifSCqRPaDr5sw&hl=es&ei=Y98tTMa7NuL9sQbH8fi1Ag&sa=X&oi=book_result&ct=result&resnum=10&ved=0CDUQ6AEwCTgU#v=onepage&q=refsum%20enfermedad%20enzima&f=false. Consultado el 1 de enero de 2011.
- ↑ Universidad Francisco Marroquín. «Síndrome de Zellweger» (en español). Consultado el 1 de enero de 2011.
- ↑ a b Muñóz, Mapi (16 de enero de 2007). «Investigadores de la Fe y científicos alemanes detectan un nuevo gen implicado en el síndrome de Usher» (en español) (Nota de prensa). Consultado el 27 de diciembre de 2010.
- ↑ Hashimoto T, Gibbs D, Lillo C, Azarian SM, Legacki E, Zhang XM, Yang XJ, Williams DS (2007). «Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B». Gene Therapy 14 (7): pp. 584–594. doi:. PMID 17268537.
- ↑ «Usherin precursor -Homo sapiens» (en inglés). The UniProt Knowledgebase. Consultado el 27 de enero de 2011.
- ↑ «G-protein coupled receptor 98 - Homo sapiens» (en inglés). The UniProt Knowledgebase. Consultado el 27 de enero de 2011.
- ↑ Ramírez, Beatriz (2004). «CNTF: una citokina con potenciales efectos terapéuticos en patologías neurodegenerativas». Clínica y ciencia 2 (2): pp. 55-61. http://www.scribd.com/doc/24337865/Factor-Neurotrofico-Ciliar.
- ↑ Lincoln, RI (26 de marzo de 2009). «Positive Results from Neurotech's NT-501 Phase 2 Dry AMD (Geographic Atrophy) Study Demonstrate Proof of Concept» (en inglés) (Nota de prensa). Consultado el 27 de diciembre de 2010.
- ↑ Coghlan, Andy (2005). «Gene therapy is first deafness 'cure'». New Scientist (Health). http://www.newscientist.com/article/dn7003.
- ↑ Graham-Rowe D (2004). «Fetal tissue restores lost sight». New Scientist (Health). http://www.medicalnewstoday.com/articles/15535.php.
Categorías:- Sordoceguera
- Síndromes
- Enfermedades hereditarias
- Enfermedades otorrinolaringológicas
- Patologías del sistema visual
- Enfermedades raras
- Enfermedades epónimas

Wikimedia foundation. 2010.