- Deficiencia de alfa-1 antitripsina
-
Deficiencia de alfa-1 antitripsina
Deficiencia de alfa-1 antitripsina
Clasificación y recursos externos Aviso médico
Aviso médico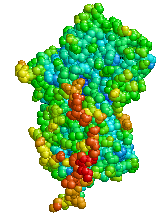
Estructura de la alfa-1 antitripsina CIE-10 E88.0 CIE-9 273.4 OMIM 107400 Medline Buscar en Medline (en inglés) MedlinePlus 000120 eMedicine med/108 MeSH D019896 Sinónimos Deficiencia de AAT. La deficiencia de alfa-1 antitripsina (abreviadamente, alfa-1 y DAAT) es un trastorno genético hereditario que puede ocasionar en la tercera y cuarta década de vida una enfermedad pulmonar obstructiva crónica (EPOC), fundamentalmente enfisema. Con menos frecuencia se puede manifestar desde el nacimiento hasta cualquier momento en la vida como una enfermedad hepática crónica. El defcit de alfa-1 se caracteriza por unos niveles en la sangre muy bajos o inexistentes de una proteína llamada alfa-1 antitripsina (AAT), que es producida por el hígado. La función principal de la AAT es proteger el tejido pulmonar de la inflamación ocasionada por las infecciones y los irritantes inhalados, como el humo de cigarrillo.
Contenido
Patofisiología
Los bajos niveles de AAT en sangre ocurren porque el hígado no puede liberar la AAT defectuosa a la rapidez normal. En un por ciento pequeño de los afectados, la acumulación de la AAT ocasiona daño grave al hígado. El defecto genético con déficit de la alfa-1 antitripsina altera la configuración de la molécula de AAT e impide su liberación por los hepatocitos. Como resultado, los niveles séricos de AAT están disminuidos, conduciendo a bajas concentraciones en el alvéolo pulmonar, donde la molécula de AAT normalmente sirve como protección como antiproteasa. Como consecuencia del exceso de proteasas las paredes alveolares son destruidas y causa enfisema.
Historia
La DAAT fue descubierta en 1963 por Carl-Bertil Laurell (1919–2001), en la Universidad de Lund, Suecia[1] , junto con el médico residente, Sten Eriksson, realizaron el descubrimiento después de notar la asuencia de la banda de α1 en la electroforesis de proteínas en 5 de 1.500 muestras; tres de los cinco pacientes desarrolaron enfisema.
La asociación con enfermedad hepática fue realizada 6 años después , cuando Sharp et al ddescriben DAAT en el contexto de enfermedad hepática[2]
Causas
La deficienca de alfa 1-antitripsina es una enfermedad infrecuente. El defecto genético responsable afecta a 1 entre 3.000-5.000 personas, es una de las tres enfermedades genéticas letales más frecuentes (las otras dos más frecuntes son la Fibrosis quística y el Síndrome de Down). Afortunadamente, no todos los pacientes con deficiencia de alfa 1- antitripsina desarrollan enfermedad clínicamente significativa. El gen de la AAt se trasmite por herencia de manera autosómica recesiva mediante dos alelos, uno de cada progenitor, que se expresan independientemete en los hijos al 50%. Se han identificado hasta 70 variantes de l gen. El conjunto de variantes se llama sistema Pi (protease inhibitor). La mayoría de las variantes carecen de significado clínico, hay alrededor de 30 que pueden tener repercusiones patológicas. Las variantes se clasifican de acuerdo con su velocidad de migración electroforética. El alelo normal presente en más del 90% de la población es el PiM. Los alelos deficientes más frecuentes son el PiS (expresa aproximadamente 50-60% de AAT) y el PiZ (expresa alrededor del 10-20% de AAT).
Enfermedades relacionadas con DAAT
El DAAT confiere una predisposición para desarrollar enfermedades a los largo de la vida, principalmente enfisema y diversos tipos de hepatopatías (colestasis neonatal, hepatitis juvenil, cirrosis hepáticas en niños y adultos y hepatocarcinomas). Las hepatopatías se realcionan con la acumulación intrahepática de polímeros y el enfisema con las bajas concentraciones de AAT, insuficientes para proteger el tejido conectivo del pulmón de los efectos negativos de las proteasas. Se ha sugerido, también, que el DAAT puede estar asociado al riesgo de desarrollo de cáncer, incluido de vesícula biliar, pulmón y linfoma. Existen pruebas de relación entre DAAT y vasculitis sistémicas y panículitis necrotizante.
Señales y síntomas más comunes
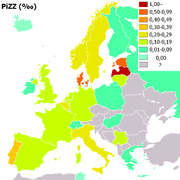 Distribución del gen PiZZ en Europa
Distribución del gen PiZZ en Europa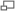
- Falta de aire en reposo o al realizar algún esfuerzo
- Fatiga o jadeo
- Tos crónica o producción crónica de flema
- Infecciones pulmonares frecuentes
- Alergias todo el año
- Rápido deterioro de la función pulmonar sin una historia significativa de tabaquismo
- Ictericia (coloración amarilla de ojos y piel)
- Acumulación de líquido en el abdomen (ascitis)
- Sangrado gastrointestinal (proveniente de las varices del estómago o esófago)
Datos importantes acerca del alfa-1
- El alfa-1 es la principal causa de trasplante de hígado en niños.
- El alfa-1 no se diagnostica con la frecuencia debida y con demasiada frecuencia se diagnostica incorrectamente como asma o como enfisema ocasionado por el fumar.
- Las personas con alfa-1 pasan un promedio de 7 años y visitan un promedio de 3 médicos antes de recibir un diagnóstico correcto.
- El 95 por ciento de las personas que se estima tienen alfa-1 no han sido diagnosticadas.
- Se estima que del 1-3 por ciento de las personas con EPOC tienen alfa-1.
- El alfa-1 ha sido identificado en virtualmente todas las poblaciones, pero es más común en personas de ascendencia del norte de Europa (Escandinavia e Inglaterra) o de Iberia (España y Portugal).
- Se estima que unos 100.000 europeos y un número similar de estadounidenses tienen alfa-1.
- Se estima que 26 millones de personas sólo en Estados Unidos desconocen que son portadores del gen anormal que ocasiona el alfa-1 y pueden pasar el gen a sus hijos. Los portadores también pueden estar en riesgo de padecer de enfermedad hepática y pulmonar sin son fumadores.
- La Organización Mundial de la Salud (reunión 1996) recomienda que todos los adultos con EPOC y todos los adolescentes y adultos con asma se hagan la prueba que mide los niveles de la proteína AAT en sangre [cita requerida].
Personas que deben considerar someterse a una prueba de detección
En octubre de 2003 la Sociedad Torácica Americana y la Sociedad Respiratoria Europea emitieron conjuntamente las nuevas guías para el diagnóstico y tratamiento del Alfa-1, publicadas en la revista médica, American Journal of Respiratory and Critical Care Medicine. Se recomienda que consideren hacerse la prueba que detecta el Alfa-1 todas las personas con las siguientes condiciones:
- EPOC — enfisema y bronquitis crónica
- Asma con obstrucción de flujo de aire que no revierte completamente con un tratamiento agresivo con broncodilatadores
- Bronquiectasias
- Enfermedad hepática crónica sin causa aparente
- Antecedentes familiares de EPOC, bronquiectasias, enfermedad hepática, alfa-1
- Parejas de personas diagnosticadas con alfa-1
- Personas con paniculitis o vasculitis C-ANCA positivo
Importancia de hacerse la prueba
En los afectados de alfa-1 el fumar es el principal factor de riesgo para el desarrollo precoz de EPOC. Otros factores de riesgo incluyen las infecciones pulmonares, el asma y la contaminación ambiental. La detección temprana del alfa-1 es importante para que los afectados porque existen medidas preventivas y de tratamiento que podrían detener la pérdida de función pulmonar y de calidad de vida. Para los fumadores afectados el dejar de fumar es la medida más importante. Hoy en día existen tratamientos para detener el daño pulmonar y para tratar los síntomas pulmonares del alfa-1.
Por su carácter hereditario (puede pasarse el trastorno a sus hijos) es importante al momento de tomar decisiones relacionadas con la reproducción de parejas.
Una sencilla prueba de sangre es todo lo que se necesita para detectar el Alfa-1.
Referencias
- ↑ Laurell CB, Eriksson S (1963). «The electrophoretic alpha 1-globulin pattern of serum in alpha 1-antitrypsin deficiency» Scand J Clin Lab Invest. Vol. 15. pp. 132–40.
- ↑ Sharp H, Bridges R, Krivit W, Freier E (1969). «Cirrhosis associated with alpha-1-antitrypsin deficiency: a previously unrecognized inherited disorder.» J Lab Clin Med. Vol. 73. n.º 6. pp. 934-9. PMID 4182334.
Fuentes
Enlaces externos
Categorías: Enfermedades hereditarias | Enfermedades metabólicas | Hepatología
Wikimedia foundation. 2010.