- Síndrome de von Hippel-Lindau
-
Síndrome de Von Hippel-Lindau Clasificación y recursos externos CIE-10 Q85.8 CIE-9 759.6 OMIM 193300 eMedicine ped/2417 Sinónimos Enfermedad de von Hippel Lindau.  Aviso médico
Aviso médico 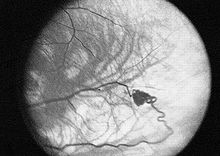
El Síndrome de von Hippel-Lindau, también conocida por sus sinónimos, Angiomatosis familiar cerebeloretinal, hemangioblastomatosis o angiofacomatosis retiniana y cerebelosa, consiste en el crecimiento anormal de vasos retiniano-cerebelosos, y se cataloga como una enfermedad rara de carácter hereditario autosómico dominante, dentro del grupo de las facomatosis. La enfermedad fue descrita por dos grupos independientes, dirigidos por Eugen von Hippel (en 1894) y Arvid Lindau (en 1926). La causa de la enfermedad es la mutación de ambos alelos del grupo VHL, causada en el uno por factores genéticos y en el segundo tras una mutación de novo. El síndrome se caracteriza por aumentar la predisposición a los tumores de riñón, del sistema nervioso central —en particular el cerebelo— y por afectar a la retina. No existe por el momento un tratamiento médico de cura, pero el conocimiento de su sintomatología y la investigación genética posibilitan que actualmente sea posible establecer diagnósticos precoces antes de la aparición de las complicaciones derivadas de la proliferación de tumores.
Epidemiología
Un estudio realizado en Inglaterra cifra la frecuencia de esta alteración heterozigótica en torno a 1:53.000, es decir 1:36.000 nacimientos.[1] Otra investigación realizada en Alemania estimó la frecuencia de la enfermedad en 1:38.000.[2] Se calcula que en Polonia viven alrededor de 1000 personas afectadas del síndrome de Hippel-Lindau.[3]
Etiología
La enfermedad se manifiesta mediante un patrón autosómico dominante. Está relacionada con una mutación genética en la síntesis del gen supresor tumoral de von Hippel-Lindau (VHL), situado en el cromosoma 3. Según establece la hipótesis de Knudson, para que se exprese el fenotipo, es necesario que el individuo sea previamente portador de la mutación en uno de los alelos, y posteriormente la mutación somática del segundo. A este mecanismo se le conoce como pérdida de heterocigosidad (abreviado como LOH, por sus siglas en inglés: loss of heterozygosity).[4] En la mayoría de los afectados por el síndrome (80%), la mutación constitutiva del gen VHL es heredada del progenitor, y la mutación de novo es la responsable del 20% de los casos de la enfermedad de von Hippel-Lindau. Se han descrito casos de mosaicismo parecental, pero su incidencia no es conocida. La descendencia de un individuo con VHL posee una riesgo del 50% de heredar la mutación causante de la enfermedad. Las pruebas prenatales en los embarazos en riesgo es posible si la mutación causante de la enfermedad ha sido identificada previamente en un miembro de la familia.[5] Muy raramente, la mutación se origina en la etapa embrionaria. En ese momento la presencia del gen es sólo parcial en el embrión aún no formado (mosaicismo).[6] Se ha probado que el polimorfismo en el gen de la ciclina D1 (CCND1) en el locus 11q13 puede verse modificado en fenotipos afectados por la mutación del gen VHL.[7]
Penetrancia genética
Las mutaciones causantes de VHL son muy penetrantes. Casi todas las personas que tienen una mutación en el gen VHL expresarán síntomas relacionados con la enfermedad para la edad de los 65 años.
Patofisiología
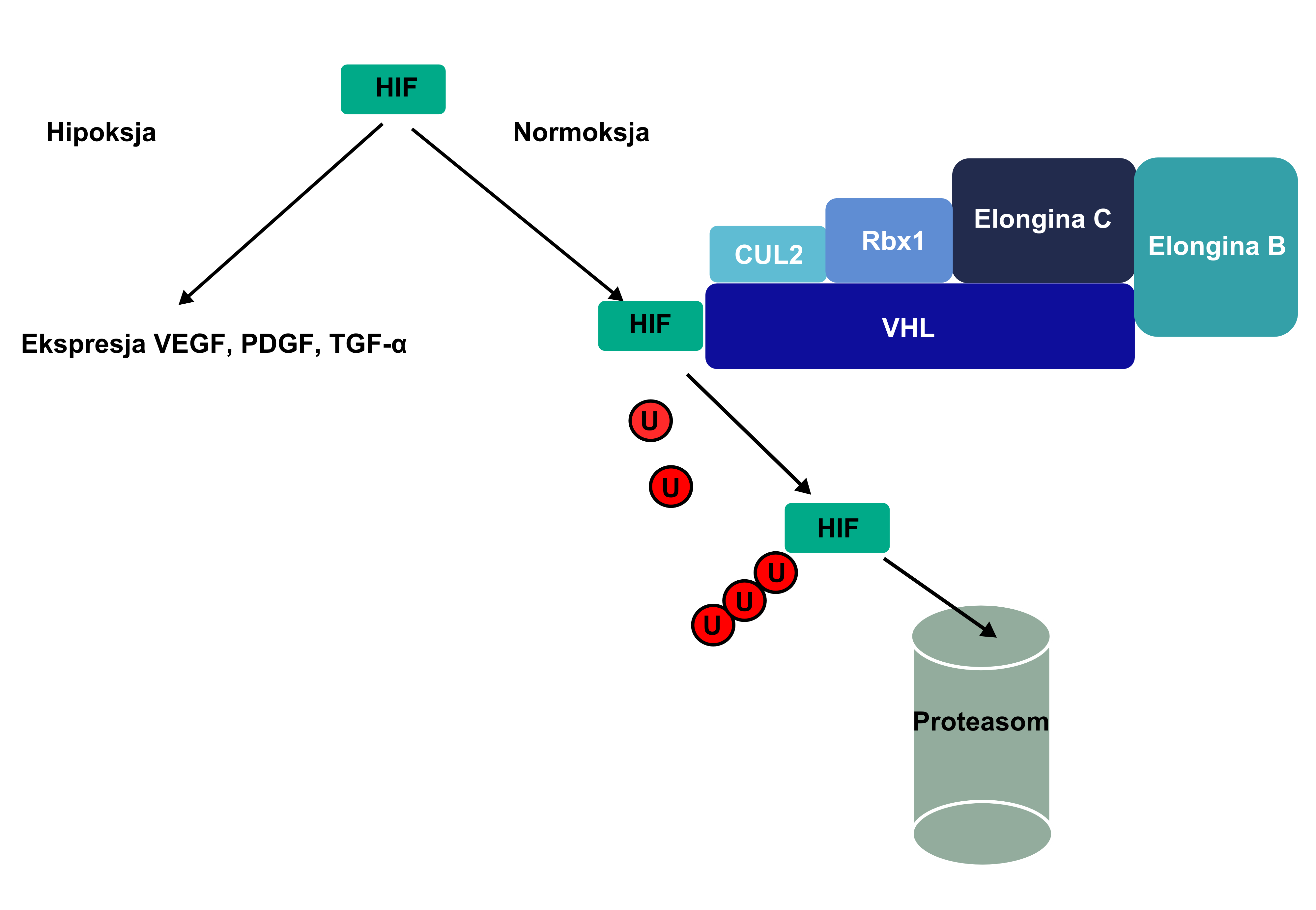
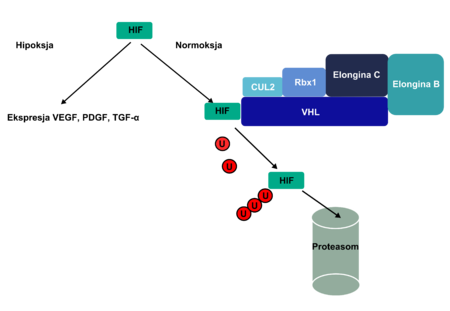 Esquema que presenta el funcionamiento del complejo VHL-elongina B-elongina C (VBC). La "U" representa la ubicvitina.
Esquema que presenta el funcionamiento del complejo VHL-elongina B-elongina C (VBC). La "U" representa la ubicvitina.
El producto de la proteína clara del gen VHL tiene una longitud de 213 aminoácidos y su funcionamiento se relaciona con la proteína clara elongina-B y elongina-C. Esta relación en el complejo VHL-elongina B-elongina C mantiene la ligazón entre las proteínas claras especificadas y es la base de su ubicuitinación (el complejo tiene un nivel de estructura proteínica E3). Se afirmó que los sustratos del complejo VBC son las proteínas claras HIF1α y HIF2α, junto a la proteína clara atípica λ (de kinaza??); cuando esta ligadura tiene lugar se establece la proteína clara domena β VHL; por ello la proteínas claras quedan sujeta a ubicuitinación y determinadas por la misma, degradándose a proteasomas.
Síntomas y desarrollo
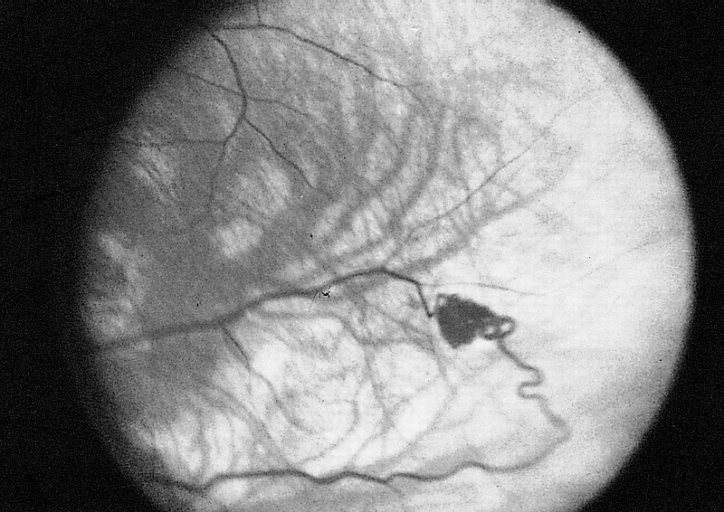
En los pacientes del síndrome de von Hippel-Lindau se observa la proliferación de tumores, de los cuales lo más esencial -desde el punto de vista clínico- es su localización en el cerebro, la médula espinal, la retina y los riñones. Aparte de esto, muchas otras alteraciones clínicas pueden afectar a otros órganos y sistemas. Los tumores que responden al síndrome de von Hippel-Lindau se caracterizan, por lo general, por sus múltiples focos, y del mismo modo se multiplican más rápidamente en un paciente joven que en la población de edad más avanzada.
Rasgos
Los rasgos del VHL son:
- Angiomatosis - pequeños nodulos de capilares en la retina y otros órganos.
- Hemangioblastomas - tumores del sistema nervioso central (especialmente en el cerebelo, tronco del encéfalo y médula espinal).
- Feocromocitoma - tumores de la médula adrenal que frecuentemente producen catecolaminas.
- Carcinoma de células renales - tumores malignos del riñón.
- Páncreas - cistos y tumores del páncreas, que pueden ser tumores neuroendocrinos.
Sin tratamiento, el VHL puede provocar ceguera y daño cerebral permanente. El fallecimiento sobreviene habitualmente por complicaciones de los tumores cerebrales o renales y enfermedades cardiovasculares secundarias al feocromocitoma. Con una detección temprana y el tratamiento apropiado, hoy en día hay más esperanzas que nunca para las personas con VHL.
Pronóstico
El pronóstico de los pacientes con la enfermedad de von Hippel-Lindau está directamente relacionado con el conocimiento de los antecedentes genéticos familiares, y con la frecuencia de exámenes periódicos personales. Ya que cerca de un 97% de los casos cuentan con antecedentes familiares conocidos, es posible establecer un diagnóstico temprano que se basa en el historial genético familiar. En los estudios realizados a gran escala con grupos de afectados por el síndrome en el sureste de Inglaterra, la causa de muerte más común se debía a complicaciones vasculares en el cerebelo (47,7%), y la esperanza de vida rondaba los 41 años.[8]
Diagnóstico clínico
El diagnóstico clínico de la enfermedad de von Hippel-Lindau (VHL) se establece en:
- Un caso simple (es decir, un individuo sin antecedentes familiares de VHL) que presenta dos o más lesiones características (por ejemplo, dos o más hemangioblastomas de la retina o el cerebro o un hemangioblastoma único asociado con una manifestación visceral como quistes del riñón o páncreas; carcinoma de células renales, o feocromacitomas suprarrenales, y, con menor frecuencia, los tumores del saco endolinfático, cistoadenomas papilares del epidídimo o del ligamento ancho, o tumores neuroendocrinos del páncreas).
- Un individuo con un historial familiar positivo del síndrome de VHL, en los que uno o más de las siguientes manifestaciones de la enfermedad está presente: angioma retiniano, espinal o hemangioblastoma cerebeloso, feocromocitoma, múltiples quistes pancreáticos, epidídimo o citoadenomas, múltiples quistes renales, o carcinoma de células renales anes de la edad de 60 años.
Pruebas de genética molecular
El gen VHL es el único gen conocido asociado con el síndrome VHL. Para detectar la presencia de mutaciones en este gen que puedan dar lugar a la enfermedad se realizan actualmente las siguientes pruebas clínicas:
- Análisis de la secuencia. Se analiza la secuencia genética de los tres exones constituyentes del gen para detectar posibles mutaciones puntuales y pequeñas deleciones o inserciones en el gen VHL. Estas alteraciones representan aproximadamente el 72% de las mutaciones de VHL.
- Análisis de supresión/duplicación. Pruebas como FISH, PCR cuantitativa, PCR a tiempo real... permiten detectar duplicaciones/deleciones completas o parcioles del gen VHL que no son fácilmente detectable por los análisis de la secuencia de ADN genómico.
Pruebas prenatales
Es posible el diagnóstico prenatal de la enfermedad para los embarazos con un 50% de riesgo de padecer VHL. Para ello se realiza el análisis del ADN extraído de las céulas fetales obtenidas por amniocentesis (generalmente se realiza entre las semanas 15 y 18 de gestación) o sobre muestreo de vellosidades coriónicas (CVS) (realizado alrededos de la semana 10 a 12 de gestación). El alelo causante de la enfermedad de un miembro de la familia afectado debe ser identificado antes de la prueba prenatal para ganarantizar la interpretación correcta del resultado de la prueba.
- NOTA: la edad gestacional se expresa como semanas menstruales o calculadas a partir del día primero del último período menstrual normal, o mediante mediciones de ultrasonido.
Las solicitudes de pruebas prenatales para síndromes como el que aquí nos ocupa, que no afectan al intelecto y tienen tratamiento disponibles no son muy comunes. Sin embargo, pueden existir diferencias de perspectivas entre los médicos y familias, especialmente si la prueba está siendo considerada para realizar una interrupción del embarazo en caso positivo. Por ello, es necesario aportar toda la información a la familia para que ésta tome la decisión que considere oportuna.
Diagnóstico genético preimplantacional (DGP)
Utilizado con éxito en embarazos que por antecedentes familiares presentan un alto riesgo para la enfermedad. Están disponibles para las familias en las que la mutación causante de la enfermedad ha sido identificada.
Historia
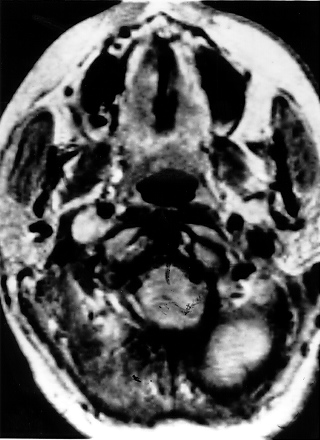
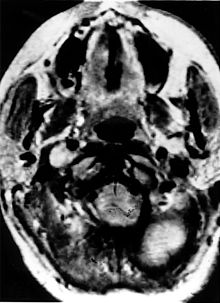 Imagen de tomografía axial computarizada con refuerzo por contraste de un paciente con cáncer bilateral de riñón producido por el síndrome de von Hippel-Lindau
Imagen de tomografía axial computarizada con refuerzo por contraste de un paciente con cáncer bilateral de riñón producido por el síndrome de von Hippel-LindauEugen von Hippel fue quien primero describió los angiomas oculares (1904),[9] y Arvid Lindau describió los angiomas del cerebelo y la espina dorsal en 1927.[10]
En un artículo que apareció en Associated Press, los endocrinólogos de la Universidad Vanderbilt que la hostilidad subyacente a la reyerta de Hatield-MacCoy podría haber sido debida en parte a las consecuencias del Síndrome de von Hippel-Lindau. El artículo sugiere que la familia McCoy tenía predisposición al mal temperamento porque muchos de ellos padecían feocromocitoma, que producía un exceso de adrenalina y tendencia hacia un temperamento explosivo.[11] Los feocromocitomas producen oleadas de adrenalina que son frecuentemente percibidas como ataques de pánico y de ira. Sin tratamiento, pueden provocar serios problemas cardiovasculares, ataques al corazón y accidentes cerebrovasculares. Solo el 20% de las personas con VHL desarrollan feocromocitomas.[12]
Tipos
Hay varios subtipos:
- Tipo 1 (angiomatosis sin feocromocitoma)
- Tipo 2 (angiomatosis con feocromocitoma)
- Tipo 2A (riesgo bajo de carcinoma de células renales)
- Tipo 2B (alto riesgo de carcinoma de células renales)
- Tipo 2C (Sólo feocromocitoma sin angiomatosis ni carcinoma de células renales)
Genética
La causa de la enfermedad es una mutación que afecta al gen oncosupresor VHL, el cual se encuentra en el brazo corto del cromosoma 3.
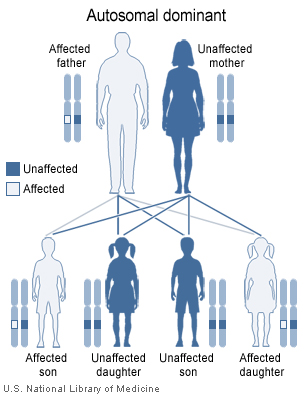
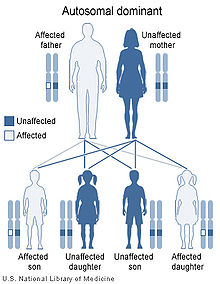 El síndrome de von Hippel-Lindau se hereda con un patrón autosómico dominante
El síndrome de von Hippel-Lindau se hereda con un patrón autosómico dominante.
Existe una amplia variedad en cuanto a la edad de inicio de la enfermedad, el órgano afectado y la gravedad del efecto. La mayor parte de los afectados del síndrome de von Hippel-Lindau heredan una copia alterada del gen, de uno de los progenitores. No obstante, en aproximadamente un 20% de los casos el gen alterado es el resultado de una nueva mutación que se da durante la formación de las células germinales o en el desarrollo embrionario.
Puesto que una copia del gen VHL normal produce una proteína funcional (pVHL), no se forman tumores. Pero si se produce una mutación en la segunda copia del gen a lo largo de la vida del afectado, la célula se quedará sin copias funcionales y no producirá la proteína VHL. La carencia de esta proteína permitirá desarrollarse a los tumores típicos del síndrome.
Sinonimia
Otros nombres son: Angiomatosis cerebelorretiniana, angiofacomatosis de retina y cerebelo, angiomatosis familiar, hemangioblastomatosis cerebelo-retiniana y enfermedad de von Hippel.
Referencias
- ↑ Maher ER, L. Iselius, JRW Yates, M. Littler, C. Benjamin, R. Harris, J. Sampson, A. Williams, M.A. Ferguson-Smith, N. Morton; "Von Hippel-Lindau disease: a genetic study", en J Med Genet, nº 28, págs. 443-447, 1991. (en inglés).
- ↑ HPH. Neumann y O.D. Wiestler; "Clustering of features of von Hippel-Lindau syndrome: evidence for a complex genetic locus", en Lancet,nº 337, págs. 1052-1054, 1991. (en inglés).
- ↑ C. Cybulski, K. Krzystolik, J. Lubiński; "Nowotwory dziedziczne 2002. Profilaktyka, diagnostyka, leczenie", ed. Termedia Wydawnictwa Medyczne, Poznań (Polonia) 2000.
- ↑ «Mutation and cancer: statistical study of retinoblastoma» (en inglés). PNAS (Proc Natl Acad Sci USA) 68 (4): pp. 80-223. 1971.
- ↑ F.M. Richards, S.J. Payne, B. Zbar, N.A. Affara, M.A. Ferguson-Smith, E.R. Maher. «Molecular analysis of de novo germline mutations in the von Hippel-Lindau disease gene» (en inglés). Hum Mol Genet (4): pp. 2139-2143.
- ↑ M.T. Sgambati, C. Stolle, P.L. Choyke, M.M. Walther, S. Pack, W.M. Lineham, G.M. Glenn (2000). «Mosaicism in von Hippel-Lindau disease: lessons from kindreds with germline mutations identified in offspring with mosaic parents» (en inglés). Am. J. Hum. Genet 66 (1): pp. 84-91. PMID 10631138.
- ↑ M- Zatyka, N.F. da Silva, S.C. Clifford, M.R. Morris, M.S. Wiesener, K.U. Eckardt, R.S. Houlston, F.M. Richards, F. Latif, E.R. Maher. «Identification of cyclin D1 and other novel targets for the von Hippel-Lindau tumor suppressor gene by expression array analysis and investigation of cyclin D1 genotype as a modifier in von Hippel-Lindau disease» (en inglés). Cancer Res 62 (13): pp. 3803-3811. http://cancerres.aacrjournals.org/cgi/content/full/62/13/3803.
- ↑ I.R. Maddock, A. Moran, E.R. Maher, A. Norman, S.J. Payne, R. Whitehouse, C. Dodd, M. Lavin, N. Hartley, et al.; "A genetic register for von Hippel-Lindau disease", en J Med Genet, nº 33, págs. 120-127
- ↑ Von Hippel E. Ueber eine sehr seltene Erkrankung der Netzhaut. Albrecht von Graefes Arch Ophthal 1904;59:83-106.
- ↑ Lindau A. Zur Frage der Angiomatosis Retinae und Ihrer Hirncomplikation. Acta Ophthal 1927;4:193-226.
- ↑ Hatfield-McCoy feud blamed on ‘rage’ disease. MSNBC.com. 2007-04-05. http://www.msnbc.msn.com/id/17967965/. Consultado el 05-04-2007.
- ↑ 'Pheochromocytoma Information'. vhl.org. 2007-04-05. http://www.vhl.org/pheo. Consultado el 05-04-2007.
Enlaces externos
Categorías:- Síndromes
- Enfermedades hereditarias
- Enfermedades raras
- Páncreas
Wikimedia foundation. 2010.