- Mutación y síndrome
-
El presente artículo enumera ciertos síndromes causados por mutación genética. Dicha enumeración depende permanentemente del estado de actualidad de la investigación médica y no puede por tanto en ningún caso tratarse de una enumeración exhaustiva.
Mutación
Una mutación es un cambio en la secuencia o en la organización del ADN.
Categorías de las mutaciones humanas
Las mutaciones pueden clasificarse en: Mutaciones genómicas, que modifican el número de cromosomas de una célula, son aneuploidías producidas por fallos durante la segregación de cromosomas durante las fases de división; mutaciones cromosómicas, que afectan a un solo cromosoma provocando duplicaciones parciales de forma espontánea o provocada por un agente mutágeno y mutaciones génicas, que alteran genes concretos afectando a un solo par de bases o a millones de ellas. Todos esos tipos de mutaciones pueden darse tanto en ADN nuclear como mitocondrial o cloroplastidial; en el caso del ADN nuclear, sólo las mutaciones ocurridas en los gametos serán transmitidas a la descendencia, las somáticas sólo producirán un cierto grado de mosaicismo en el individuo que las porte. Las mutaciones pueden provocar diversas enfermedades, como por ejemplo los síndromes genéticos. Es destacable que una pequeña mutación puede alterar toda una proteína y por consiguiente, su función en el organismo. En cambio, otros cambios en el genotipo de un individuo no provocan ningún efecto notable, ya que la mutación proiducida puede haber afectado al ADN en un segmento que no se a crítico para el organismo o por que el cambio no altere la secuencia de aminoácidos o incluso que el aminoácido mutado no modifique las propiedades de la proteína. No todas las mutaciones tienen consecuencias clínicas, pero conocer las mutaciones tiene una doble utilidad, por una parte conciencian de la fragilidad humana en sí y la de su herencia y por otra, permite diagnosticar diversas enfermedades desde un punto de vista genético y bioquímico.
Origen de las mutaciones
Mutaciones genómicas
Las mutaciones genómicas son las que provocan las aneuploidías cromosómicas y las más frecuentes en el ser humano. Se producen a raíz de un error el la disyunción durante la meiosis en una tasa bastante baja, aunque debe ser mayor si se puediesen contar todos los embarazos de fetos aneuploides que terminan en aborto espontáneo antes de que se les detecte la aneupliodía.

Mutaciones cromosómicas
Las mutaciones cromosómicas son más comunes que las genómicas y normalmente no son transmitidas a la descendencia porque sobrevivir con una de estas mutaciones es casi imposible.
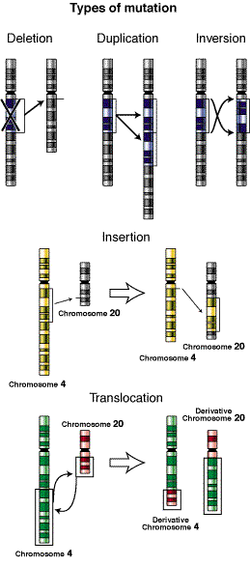 Tipos de mutaciones cromosómicas.
Tipos de mutaciones cromosómicas.
Mutaciones génicas
Son las sustituciones de pares de bases en las inserciones y las deleciones que se originan por: errores producidos en el proceso de replicación del ADN o durante la reparación del ADN. Algunas mutaciones son espontáneas mientras que otras son inducidas mediante agentes físicos o químicos denominados mutágenos.
Errores en la replicación del ADN
La mayor parte de los errores de la replicación son eliminados y corregidos por enzimas que reconocen la secuencia incorrecta y sustituyen las bases, todo durante la corrección de pruebas. La tasa de error el de un nucleótido por cada diez mil introducidos y la de corrección es del 99,9%, por tanto, la tasa total de mutaciones por replicación es una cifra muy pequeña.
Reparación de daño del ADN
Aproximadamente diez mil y un millón de nucleótidos por célula humana y día sufren mutaciones químicas espontáneas como depurinaciones, desmetilaciones y desaminaciones, debidas en gran parte a reacciones con mutágenos químicos del ambiente y por exposición a radiaciones ionizantes o ultravioleta. Las bases son reparadas normalmente, pero no la totalidad; además es posible que el mecanismo de reparación no lea con exactitud la cadena molde y provoque mutaciones.
Tipos de mutaciones y sus consecuencias
Sustituciones de nucleótidos
Mutaciones de cambio de sentido
La sustitución de un único nucleótido,o sea, una mutación puntual en una secuencia de ADN, puede alterar un codón y causar la sustitución de un aminoácido por otro. Éstas son las mutaciones de cambio de sentido que alteran el significado del gen al especificar un aminoácido diferente. En trastornos como las hemoglobinopatías, la mayoría de las mutaciones de cambio de sentido; ciertas mutaciones en la región 5’ del promotor o en la región 3’ no traducida del gen de la β-globina conducen a un descenso pronunciado de la cantidad producida de mRNA de la β-globina procesado y maduro.
Mutaciones sin sentido
Las sustituciones puntuales en la secuencia de ADN que causan la sustitución de un codón normal por un codón de parada (stop codon), son las mutaciones sin sentido y generan una terminación prematura de la traducción.La traducción del mRNA cesa al alcanzar un codón de terminación, una mutación que convierte una secuencia codificante, presente en un exón, en un codón de terminación ocasiona que la traducción se detenga a medio camino de la secuencia codificante del mRNA. Las consecuencias son dos: En primer lugar, el mRNA portador de una mutación prematura es a menudo inestable, se degrada y se imposibilita la traducción, incluso si el mRNA es estable, la proteína traducida no suele serlo y es rápidamente degradada en la célula; en segundo lugar, una mutación puntual puede destruir el codón de terminación normal y permitir que la traducción continúe hasta alcanzar el codón de terminación siguiente; se crea así una proteína con aminoácidos adicionales en su extremo carboxilo que puede alterar toda función de regulación ejercida por la región 3’ no traducida justo a partir del codón de terminación normal.
Mutaciones durante el procesamiento del ADN
La maduración del RNA a mRNA maduro requiere una serie de modificaciones: la adicción de la caperuza en 5’, la poliadenilación y la eliminación de intrones. Hay dos tipos de mutaciones relacionadas con estos procesos: las que afectan a bases necesarias que interfieren o impiden el proceso de corte y empalme o las que implican una sustitución de bases del intrón. Esta variedad, crea sitios de corte y empalme alternativos que provocan que secuencias de ADN sean ensambladas de forma incorrecta.
Puntos críticos de mutación
Los cambios de nucleótidos que conllevan la sustitución de una purina por otra o de pirimidina por otra se denominan “transiciones”. La sustitución de una purina por una pirimidina o viceversa se llama “transversión”. Si las sustituciones de nucleótidos se produjesen al azar, habría el doble de transversiones que de transiciones, porque cada base puede sufrir dos tranversiones pero sólo una transición. Sin embargo, diferentes procesos mutagénicos causan preferentemente uno u otro tipo de sustitución. Por ejemplo, las transiciones se encuentran principalmente en las sustituciones de un solo par de bases que originan enfermedades génicas. La explicación para este fenómeno es probablemente, que la principal forma de modificación del ADN implica la mutilación de los residuos de citosina (C), sobre todo, cuando están situados inmediatamente a una guanina; la desaminación espontanea de la 5’ metilcitosina a timidina (T) en el doblete C-G (guanina), da transiciones de C a T o de G a A (adenina). Más del 30% de todas las sustituciones de un único nucleótido son de este tipo y se producen con una tasa veinticinco veces superior a la de cualquier otra mutación que afecte a un solo nucleótido.
Deleciones e inserciones
Las mutaciones también pueden producirse por inserción, inversión, fusión y deleción de secuencias de ADN. Algunas sólo involucran a unos pocos nucleótidos y se detectan mediante el secuenciado de nucleótidos; en otros casos, un segmento grande de un gen o un gen entero se delecionan, invierten, duplican o translocan, lo que crea una nueva secuencia génica.
Mutaciones de cambio de marco de lectura
Son deleciones e inserciones pequeñas que afectan a un número pequeño de pares de bases. Si el número de bases implicadas no es tres o un múltiplo de tres, es decir, no es un número entero de codones y cuando ocurren en una secuencia codificante, el marco de lectura se altera empezando en el punto de inserción o deleción. Como resultado, se codifican aminoácidos anormales. Por el contrario, si el número de pares de bases insertadas o delecionadas es tres o múltiplo de tres, no se produce un cambio de marco y los aminoácidos correspondientes serán insertados o delecionados en el producto génico traducido.
Deleciones e inserciones grandes
Las alteraciones de la estructura génica lo suficientemente grandes como para ser detectadas por transferencia Southern son poco comunes, pero han sido descritas en muchos trastornos hereditarios. La frecuencia de estas mutaciones difiere de forma notable entre las distintas enfermedades genéticas. Por ejemplo, las deleciones en el gen de la distrofina, que es muy grande y está situado en el cromosoma X, causa la distrofia muscular de Duchenne. En algunos casos se conocen bien los fundamentos de la deleción génica, que es mediada probablemente por una recombinación aberrante entre copias múltiples de secuencias de ADN similares o idénticas. La inserción de grandes cantidades de ADN es una causa de mutación mucho más rara en la deleción.
Efectos de la recombinación
Una causa de mutación importante encontrada algunas enfermedades implica la deleción o duplicación mediadas por la recombinación entre secuencias muy similares o idénticas de ADN. En otros casos, un gen puede pertenecer a una familia génica representada por copias similares del gen situado en tándem un cromosoma. Cuando los genes de una misma familia se localizan en tándem de cabeza a cola en la misma región cromosómica, a veces se desalinean y se aparean erróneamente en la meiosis o en la mitosis después de la replicación. Una recombinación que se produzca entre cromosoma mal apareados o entre dos cromátidas hermanas puede ocasionar deleciones. Se piensa que este mecanismo es el responsable de la deleción de uno de los genes de la α-globina en la α-talasemia. También pueden producirse recombinación anormal entre dos secuencias repetidas similares en una sola cadena de ADN. Dependiendo de la orientación de estas secuencias, la recombinación puede llevar a una o una mutación.
Mutaciones dinámicas
Las mutaciones en trastornos como la enfermedad de Huntington y el síndrome del X frágil implican la amplificación de secuencias repetidas de trinucleótidos. En estas enfermedades, una simple repetición de un trinucleótido situada en la región codificante o en una región transcrita pero no traducida de un gen, puede expandirse durante la gametogénesis. Es lo que se denomina mutación dinámica e interfiere en la expresión normal del gen. Ésto ocasionará un producto proteico anormal, además la expansión de la repetición en las regiones transcritas pero no traducidas de un gen pueden interferir con la transcripción en el procesamiento del mRNA. No se comprende totalmente cómo se producen las mutaciones dinámicas; se piensa que durante la replicación se pueden producir errores cuando la cadena en crecimiento se desliza mientras la polimerasa está intentando extenderla y posteriormente regresa a la plantilla fuera del sitio en el que estaba cuando perdió contacto con la cadena molde.
Síndrome
Introducción
Toda la información sobre los caracteres, estructura y función del organismo de los seres vivos se encuentran en los genes, dentro de los cromosomas, en forma de ADN. En la fecundación, dos juegos simples de cromosomas, uno de cada progenitor, se unen para formar parte de la información genética de cada individuo; es lo que se conoce como genoma. Si durante este proceso se produce alguna mutación, puede ocurrir lo que denominamos síndrome genético. En muchos casos, el origen se debe a que uno o ambos padres son portadores de una alteración genética que es susceptible de ser transmitida a los hijos. En otros muchos casos se desconocen las causas exactas por la que un síndrome genético se da en un individuo concreto sin haber antecedentes familiares. Se debe diferenciar entre un síndrome genético, que se produce durante la embriogénesis por una mutación heredada o no, de otras patologías que pueden aparecer durante el desarrollo del feto y que también conducen a problemas del desarrollo posterior. Los síntomas más inmediatos son: bajo peso a l nacer, anomalías cardíacas,… Las alteraciones genéticas conllevan importantes variaciones en el desarrollo físico y psíquico del individuo. El diagnóstico se hace mayoritariamente con estudios cromosómicos y por fenotipos conductuales, es decir un patrón de síntomas característicos de la enfermedad. Hasta hace muy poco estas patologías eran prácticamente desconocidas. Los avances tecnológicos, la labor de muchas asociaciones y de familiares afectados, ha ido propiciando un cambio de tendencia. Una dificultad común es que no existe uniformidad en la magnitud y la presencia de síntomas, esto se debe a que la enfermedad puede presentar diversos grados de expresión.
Causas y tipos de anomalías genéticas
Las aberraciones cromosómicas se producen sin ninguna causa aún identificada, de forma aleatoria y es difícil detectar los factores de riesgo determinantes. Pueden clasificarse en varios tipos:
Anomalías estructurales
La duplicación: Una parte del cromosoma se duplica o presenta dos copias. Como resultado hay una información adicional que puede provocar que los genes implicados, no funcionen correctamente y se presenten errores en la secuencia de desarrollo del embrión.
La deleción: Una parte del cromosoma se pierde o se elimina. Según la cantidad de material perdido o alterado los síntomas serán más o menos severos.
Anomalías numéricas
Ocurren cuando en las células hay un número de cromosomas diferente al normal. Se habla de trisomías para describir la presencia anormal de 3 cromosomas en lugar del número normal correspondiente. La monosomía es la ausencia de uno de los miembros que conforman el par cromosómico. La anomalía cromosómica también puede darse por intercambio de lugar o traslocación.
Mosaicismo
Es la presencia de más de un tipo de célula (con carga cromosómica distinta) en un individuo. El mosaicismo se expresa a nivel médico en términos de porcentaje para expresar el número de células normales respecto a las alteradas.

Ejemplos de síndromes
Síndrome ATR-X
En todas las clases de talasemia-α descritas, las mutaciones en los genes de la α-globina o en sus secuencias cis explican la reducción de la síntesis en la α-globina. Por el contrario, hay un tipo de α-talasemia (el síndrome ATR-X) que se debe a mutaciones en el gen ATR X, lo que da lugar a una reducción de la actividad con expresión de una proteína de remodelación de la cromatina (la proteína ATR X) que actúa en dirección trans activando la expresión de los genes de la α-globina. Inicialmente, se consideró que el síndrome ATR-X era un trastorno específico, debido a la aparición de la enfermedad por hemoglobina H, además, todos los individuos afectados eran hombres que también sufrían cuadros graves de retraso mental ligado al cromosoma X junto a una amplia gama de otros defectos como características faciales típicas, alteraciones esqueléticas y malformaciones urogenitales. Esta diversidad de fenotipos sugiere que la proteína ATR X regula la expresión de otros numerosos genes además de los correspondientes a las α-globinas, aunque en este momento son desconocidos. A pesar de que no se conoce con precisión su mecanismo de acción, la proteína ATR X pertenece a una familia de proteínas de remodelación de la cromatina que actúan característicamente en el contexto de complejos proteicos grandes cuya función es la de producir cambios en la topología del ADN. Estas alteraciones topológicas dirigen la formación de estados nucleosómicos remodelados. Las alteraciones en los patrones de la metilación del ADN en los pacientes con el síndrome ATR-X indican que la proteína ATR X parece ser necesaria para establecer o mantener el patrón de la metilación en ciertos dominios del genoma, quizá a través de la modulación del acceso de la metilasa a sus sitios de unión. Todas las mutaciones identificadas hasta el momento en el gen ATR X son mutaciones con pérdida de función parciales.
Síndrome de Beckwith-Wiedemann
El síndrome de Beckwith-Wiedemann (BWS) (OMIM 130650) es un síndrome común en todas las etnias que suele ser esporádico, aunque en raras ocasiones puede ser heredado como un rasgo autosómico dominante. Afecta alrededor de una de cada 13700 nacidos vivos. El BWS se produce por un desequilibrio en la expresión del imprinting génico en la región p15 del cromosoma 11. Uno de los genes, transcritos pero no traducidos, codifica un factor de crecimiento semejante a la insulina, que promociona el crecimiento. Por el contrario, otro de los genes codifica un supresor del ciclo celular que limita la división y el crecimiento celular. El desequilibrio en la expresión de los genes 11p15 puede ocurrir mediante varios mecanismos: Mutaciones en el alelo materno por pérdida de expresión, provocan la pérdida de expresión del gen codificante del supresor y el incremento de la expresión del gen codificante del factor de crecimiento están causados por la isodisomía (dos cromosomas del mismo progenitor que son similares) paterna del 11p15. Como la recombinación somática que lleva a la disomía uniparental (con cromosomas que proceden de un mismo progenitor) ocurre después de la concepción, los individuos con este tipo de disomía, son mosaicos y pueden necesitar el análisis de otros tejidos, además de la sangre, para la detección de su disomía. Las características principales fenotípicas en la edad prenatal son: crecimiento excesivo, macroglosia, onfalocele, visceromegalia, tumor embrionario, hemihiperplasia, anomalías renales, citomegalia adrenocortical e hipoglucemia. El riesgo de recurrencia en los hermanos y los hijos con el síndrome es muy variable y aumentan con las técnicas de reproducción asistida. Los tratamientos consisten en intervenciones quirurgicas y control farmacológico de las enfermedades.
Síndrome del cáncer hereditario
El desarrollo del cáncer (oncogénesis) se debe a mutaciones en uno o más del elevado número de genes que regula el crecimiento celular y la muerte celular programada. Cuando el cáncer forma parte de un síndrome de cáncer hereditario, la mutación inicial que da lugar a la neoplasia se hereda a través de la línea de células germinales y por tanto, ya existe en todas las células del cuerpo.
Síndrome de la displasia branquio-oto-renal
Es un síndrome autosómico dominante producido por un pleiotropismo. Consiste en alteraciones en el desarrollo coclear y del oído externo, quistes y fístulas en el cuello, displasia renal y malformaciones del túbulo colector renal. Este síndrome se debe a la mutación en el gen EYA 1, que codifica una proteína fosfatasa.
Síndrome del ojo de gato
Se produce por tener un complemento cuádruple del segmento 22 q11.2 del cromosoma 22. Se caracteriza clínicamente por un coloboma ocular, defectos cardíacos congénitos, anomalías craneofaciales y un retraso mental moderado. El cariotipo en el síndrome del ojo de gato es 47,XX o XY, +inv dup (22)(pter -> q11.2).
Síndrome del carcinoma vasocelular nevoide o síndrome de Gorlin
Provoca malformaciones craneofaciales, polidactilia ocasional y quistes dentarios. Se producen por mutaciones en el gen PTCH1.
Síndrome del maullido de gato o cri du chat
Es un síndrome en el que se produce una deleción terminal o intersticial del brazo corto del cromosoma 5. El nombre lo recibe del llanto de los niños que sufren este trastorno, parecido al maullido de un gato. Las características fenotípicas son microcefalia, hipertelorismo, pliegues epicánticos, orejas de implantación baja, a veces con apéndices preauriculares y micrognatia. Otros problemas son el retraso mental grave y las malformaciones cardíacas. La mayoría de los casos son esporádicos y del 10 al 15% son hijos de portadores de una translocación. Los puntos de rotura y extensión del segmento delecionado varían, pero la región crítica se ha localizado en la banda 5p15. Se ha empezado a determinar la relación entre la monosomía de estos genes y el fenotipo clínico; muchos parecen ser debidos a haploinsuficiencias.

Síndrome linfoproliferativo autoinmunitario
Es un trastorno autosómico dominante poco frecuente que se caracteriza por una linfoadenopatía masiva y esplenomegalia, sobre todo durante la niñez. Se producen fenómenos autoinmunitarios como trombocitopenia y anemia hemolítica. La alteración primaria radica en el mecanismo de apoptosis (muerte celular natural en la que aparecen repentinamente aperturas en las membranas mitocondriales, que provoca la salida del contenido de las mismas y por consiguiente, la inactivación de las proteasas intracelulares, la fragmentación del ADN y la muerte celular) de los linfocitos mediada por el receptor Fas y su ligando. Las mutaciones negativas dominantes en un alelo de cualquier cromosoma que codifique una de estas moléculas causan la pérdida de la función receptor – ligando, lo que provoca deficiencias en las señales de apoptosis y una expansión masiva de linfocitos T inmaduros. No se conoce con exactitud el mecanismo por el que éste efecto puede incrementar los distintos tipos de linfomas.
Síndrome de Marfan
Los pacientes con síndrome de Marfan (OMIM 154700) evidencian muchas de las características de un patrón de herencia dominante, debido a su pleitropismo, múltiples aparatos y sistemas quedan dañados; posee expresividad variable. Se distingue por alteraciones del crecimiento de los huesos y tejidos de sostén, es notoria una talla alta de estatura. El crecimiento de los huesos largos y de los dedos largos y afilados produce la llamada aracnodactilia. La longitud de éstos permite que el pulgar y el meñique puedan tocarse al realizar una circunferencia en la muñeca. Lo cual se conoce como signo de la muñeca o de Walker – Murdoch. El crecimiento oseo también se relaciona con torsión de la columna vertebral y presencia de escoliosis. Por lo general se identifica durante la infancia y empeora en la adolescencia, la escoliosis, por ejemplo, es una complicación importante que requiere tratamiento a largo plazo. Debido a la laxitud articular, hay inestabilidad articular, luxaciones y debilidad, sobre todo en las articulaciones que soportan mas peso. Existen también complicaciones a nivel ocular, porque se produce la luxación de las fíbras de la zónula que sostienen al cristalino. Las características mas peligrosas, se relacionan con complicaciones en los vasos sangíneos. Las mujeres con este síndrome padecen complicaciones en el embarazo y el parto.
Estas anormalidades genéticas se presentan en el gen encargad de la proteína fibrilina del cromosoma 15. Las mutaciones producidas son variables, desde algunas mutaciones puntuales hasta deleciones e interrupción de la codificación de la proteína.
Síndrome de Wiskott – Aldrich
(OMIM 301000) Es una alteración monogénica de la función inmunitaria del gen Xp 11.2, se caracteriza por la trombocitopenia.
Síndrome de cáncer Li – Fraumeni
(OMIM 151623) Se produce una mutación alélica para el gen p53 que codifica una importante proteína cuya función es la supresión hormonal en el control de la división celular. Este síndrome constituye la explicación fisiopatológica de algunos casos de predisposicion familiar al cancer por mutaciones del alelo. Las mutaciones heredadas son muy poco frecuentes, sin embargo, las mutaciones adquiridas, son relativamente comunes (se presentan en la mitad de los tumores examinados).
Síndrome de Goodpasture
El síndrome de Goodpasture es una enfermedad autoinmunitaria, perteneciente al grupo de enfermedades de hemorragia alveolar o pulmonar, que suele acabar en una enfermedad pulmonar intersticial y se caracteriza por la producción de anticuerpos antimembrana basal tanto del glomérulo renal como de los alveolos pulmonares.
Síndrome de Kearns-Sayre
Enfermedad Neurológica Mitocondrial producida en las células del cuerpo humano con pocos casos en el mundo la misma es irreversible y aún no posee cura pero se sugiere tratamiento paliativo con medicación específica y tratamiento del dolor a los fines de evitar el fallecimiento del paciente.
Síndrome de Klinefelter
Síndrome de Angelman
Síndrome de Cornelia de Lange
Síndrome de Williams
Síndrome de von Hippel-Lindau
Síndrome de Down
Síndrome del X frágil
Síndrome del QT largo
Síndrome de Turner
Véase también
Bibliografía
- Nussbaum R.L., McInnes R. R. & Willard H. F. 2008. Thompson & Thompson Genética Médica.7ª edición. Elsevier Masson. Barcelona. 584
- Sack G. H. Jr. 2002. Genética médica. Mc Graw Hill. México. 272
- Champagny, B. 1986. Síntomas, signos y síndromes: definición y descripción. Doyma. Barcelona
- Castro del pozo, S. 1996. Manual de patología general: Etiología- fisiopatología - semiología - síndromes. Masson. Barcelona
- Klung, W. S.; Cummings, M. R. & Spencer, C. A. 2006. Conceptos de genética. 8ª edición. Pearson - Prentice Hall. Madrid. 884
Enlaces externos
Wikimedia foundation. 2010.