- Ecuación de Van der Waals
-
La ecuación de Van der Waals es una ecuación de estado de un fluido compuesto de partículas con un tamaño no despreciable y con fuerzas intermoleculares, como las fuerzas de Van der Waals. La ecuación, cuyo origen se remonta a 1873, debe su nombre a Johannes Diderik van der Waals, quien recibió el premio Nobel en 1910 por su trabajo en la ecuación de estado para gases y líquidos, la cual está basada en una modificación de la ley de los gases ideales para que se aproxime de manera más precisa al comportamiento de los gases reales al tener en cuenta su tamaño no nulo y la atracción entre sus partículas.
Contenido
Ecuación
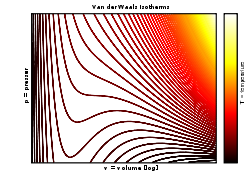 Las isotermas de Van der Waals: el modelo predice correctamente la fase de líquido prácticamente incompresible, pero las oscilaciones en la fase de transición no se corresponden con los resultados experimentales.
Las isotermas de Van der Waals: el modelo predice correctamente la fase de líquido prácticamente incompresible, pero las oscilaciones en la fase de transición no se corresponden con los resultados experimentales.
Una forma de esta ecuación es:
donde
- p es la presión del fluido
- v es el volumen en el que se encuentran las partículas dividido por el número de partículas
- k es la constante de Boltzmann
- T es la temperatura, en kelvin
- a' es un término que tiene que ver con al atracción entre partículas
- b' es el volumen medio excluido de v por cada partícula
Si se introducen el número de Avogadro, NA, el número de moles n y, consecuentemente, el número total de partículas n•NA, la ecuación queda en la forma siguiente:
donde
- p es la presión del fluido
- V es el volumen total del recipiente en que se encuentra el fluido
- a mide la atracción entre las partículas

- b es el volumen disponible de un mol de partículas

- n es el número de moles
- R es la constante universal de los gases ideales,

- T es la temperatura, en kelvin
Se debe hacerse entre una distinción cuidadosa entre el volumen disponible para una partícula y el volumen de una partícula misma. En particular, en la primera ecuación
 se refiere al espacio vacío disponible por partícula. Es decir que , es el volumen
se refiere al espacio vacío disponible por partícula. Es decir que , es el volumen  del recipiente dividido por el número total de
del recipiente dividido por el número total de  de partículas. El parámetro b', por el contrario, es proporcional al volumen ocupado de una partícula —únicamente delimitado por el radio radio atómico. Este es el volumen que se restará de debido al espacio ocupado por una partícula. En la derivación original de Van der Waals, que figura a continuación,
de partículas. El parámetro b', por el contrario, es proporcional al volumen ocupado de una partícula —únicamente delimitado por el radio radio atómico. Este es el volumen que se restará de debido al espacio ocupado por una partícula. En la derivación original de Van der Waals, que figura a continuación,  es cuatro veces el volumen disponible de la partícula. Observe además que la presión
es cuatro veces el volumen disponible de la partícula. Observe además que la presión  tiende a infinito cuando el contenedor está completamente lleno de partículas de modo que no hay espacio vacío dejado por las partículas a moverse. Esto ocurre cuando
tiende a infinito cuando el contenedor está completamente lleno de partículas de modo que no hay espacio vacío dejado por las partículas a moverse. Esto ocurre cuando  .
.Validez
Por encima de la temperatura crítica la ecuación de Van der Waals es una mejora de la ley del gas ideal, y para temperaturas más bajas la ecuación es también cualitativamente razonable para el estado líquido y estado gaseoso a baja presión. Sin embargo, el modelo Van der Waals no es adecuado para los cálculos cuantitativos rigurosos, útil restante sólo con fines educativos y de calidad.
En la transición de fase de primer orden, el rango de (P, V, T), donde la fase líquida y la fase gaseosa se encuentran en equilibrio, no lo exhibe el hecho empírico de que p es constante en función de V a una temperatura dada aunque este comportamiento se pueda insertar fácilmente en el modelo la Van der Waals, el resultado ya no es un modelo analítico simple, y otros (como los basados en el principio de estados correspondientes) logran un mejor ajuste con más o menos el mismo trabajo.
Derivación
La mayoría de los libros de texto dan dos distintas derivaciones. Una de ellas es la derivación convencional que se remonta a Van der Waals y la otra es una derivación de la mecánica estadística. Este último tiene la gran ventaja de que se hace explícito el potencial intermolecular, que se descuida en la primera derivación.
El volumen excluido por partícula es
 , que hay que dividir por dos para no contar dos veces la misma interacción, por lo que el volumen excluido del b' es
, que hay que dividir por dos para no contar dos veces la misma interacción, por lo que el volumen excluido del b' es  , que es cuatro veces el volumen adecuado de la partícula. Fue un punto de interés para Van der Waals, que el factor de cuatro es en realidad un rendimiento superior de los valores empíricos límites, b' son generalmente más bajos. Por supuesto, las moléculas no son totalmente rígidas como Van der Waals pensaba, sino con frecuencia bastante suaves.
, que es cuatro veces el volumen adecuado de la partícula. Fue un punto de interés para Van der Waals, que el factor de cuatro es en realidad un rendimiento superior de los valores empíricos límites, b' son generalmente más bajos. Por supuesto, las moléculas no son totalmente rígidas como Van der Waals pensaba, sino con frecuencia bastante suaves.A continuación, se introduce una fuerza atractiva por parejas entre las partículas. Van der Waals supone que, a pesar de la existencia de esta fuerza, de la densidad del fluido es homogéneo. Además se asume que el rango de la fuerza de atracción es tan pequeño que la gran mayoría de las partículas no sienten que el contenedor es de tamaño finito. Es decir, la mayor parte de ellas tienen más atracción a las partículas a su derecha que a su izquierda cuando están relativamente cerca de la pared de la izquierda del contenedor (y viceversa). Teniendo en cuenta la homogeneidad del líquido, la mayor parte de las partículas no experimentan una fuerza neta que tire de ellas hacia la derecha o hacia la izquierda. Esto es diferente para las partículas en las capas superficiales directamente adyacentes a los muros. Se sienten una fuerza neta de las partículas de mayor tirando de ellos hacia el recipiente, ya que esta fuerza no es compensado por las partículas en el lado donde la está pared (otro supuesto es que no hay interacción entre las paredes y las partículas, lo cual no es cierto como puede verse en el fenómeno de la formación de gotas; la mayoría líquidos muestran adhesión). Esta fuerza neta disminuye la fuerza ejercida sobre la pared por las partículas en la capa superficial. La fuerza neta sobre una partícula de la superficie, tirando de ella hacia el recipiente, es proporcional a la densidad numérica C = NA / Vm (número de partículas por unidad de volumen). El número de partículas en las capas superficiales es, de nuevo, asumiendo homogeneidad del fluido, también proporcional a la densidad. En total, la fuerza sobre las paredes se reduce por un factor proporcional al cuadrado de la densidad y la presión (fuerza por unidad de superficie) se reduce en
 ,
,
de modo que
Es de algún interés histórico señalar que Van der Waals en su conferencia del premio Nobel le dio crédito a Laplace argumentando que la presión se reduce de forma proporcional al cuadrado de la densidad.
Esto hace que el promedio de energía Helmholtz por partícula sea reducida en una cantidad proporcional a la densidad del fluido. Sin embargo, la presión obedece a la relación termodinámica
donde A*es el energía Helmholtz por partículas. La atracción, por lo tanto, reduce la presión en una cantidad proporcional a
 . Denota la constante de proporcionalidad por a, obtenemos
. Denota la constante de proporcionalidad por a, obtenemosque es la ecuación de Van der Waals.
Derivaciones convencionales
Consideremos en primer lugar un mol de gas que se compone de partículas puntuales sin interacción que satisfacen la ley de los gases ideales.
A continuación se asume que todas las partículas son esferas duras del mismo radio finito r (el radio de Van der Waals). El efecto del volumen finito de las partículas es disminuir el espacio vacío disponible en el cual se mueven libremente las partículas. Debemos reemplazar V por V − b, donde b se le llama el volumen excluido. La ecuación corregida se convierte en:
El volumen excluido b no es exactamente igual al volumen ocupado por el tamaño de sólidos, de tamaño finito, partículas, pero en realidad cuatro veces ese volumen. Para ver esto tenemos que darnos cuenta de que una partícula está rodeada por una esfera de radio r = 2r (dos veces el radio original) que está prohibido para los centros de las otras partículas. Si la distancia entre dos centros de las partículas es más pequeño que 2r, lo que significaría que las dos partículas penetran entre sí, que, por definición, las esferas duras no son capaces de hacer.
Derivación termodinámica estadística
La función de partición canónica Q de un gas ideal consistente en N = nNA partículas idénticas, es
donde Λ es la longitud de onda térmica de De Broglie,
con las definiciones usuales: H es la constante de Planck, m, la masa de una partícula, k, la constante de Boltzmann y T la temperatura absoluta. En un gas ideal
 es la función de partición de una partícula en un recipiente de volumen V. Con el fin de obtener la ecuación de Van der Waals se supone ahora que cada partícula se mueve de forma independiente en un campo de potencial promedio ofrecido por las otras partículas. El promedio más las partículas es fácil, porque suponemos que la densidad de las partículas del fluido de Van der Waals es homogénea. La interacción entre un par de partículas, que son esferas rígidas, se considera
es la función de partición de una partícula en un recipiente de volumen V. Con el fin de obtener la ecuación de Van der Waals se supone ahora que cada partícula se mueve de forma independiente en un campo de potencial promedio ofrecido por las otras partículas. El promedio más las partículas es fácil, porque suponemos que la densidad de las partículas del fluido de Van der Waals es homogénea. La interacción entre un par de partículas, que son esferas rígidas, se considerar es la distancia entre los centros de las esferas y D es la distancia que las esferas duras se tocan (el doble del radio de Van der Waals). La profundidad de la de Van der Waals es ε.
Debido a que las partículas son independientes, la función de partición total todavía se factoriza, Q = qN / N!, pero el potencial intermolecular necesita dos modificaciones a
 . En primer lugar, debido al tamaño finito de las partículas, no todos los V están disponible, pero sólo V − Nb', donde (como en la derivación convencional anterior)b' = 2πd3 / 3. En segundo lugar, introducir un factor de Boltzmann exp[ − ϕ / (2kT)] para atender el potencial intermolecular medio. Dividimos aquí el potencial de dos, porque esta energía de interacción es compartida entre dos partículas. Así
. En primer lugar, debido al tamaño finito de las partículas, no todos los V están disponible, pero sólo V − Nb', donde (como en la derivación convencional anterior)b' = 2πd3 / 3. En segundo lugar, introducir un factor de Boltzmann exp[ − ϕ / (2kT)] para atender el potencial intermolecular medio. Dividimos aquí el potencial de dos, porque esta energía de interacción es compartida entre dos partículas. AsíToda la atracción que es ejercida sobre una partícula es
donde se asume que en una capa de espesor dr hay N/V 4π r2dr partículas. Esta es una aproximación del campo medio, la posición de las partículas es un promedio. En realidad, la densidad cercana de la partícula es diferente que la que están lejanas, como pueden ser descritas por una función de correlación par. Además, se descuida que el fluido está encerrado entre las paredes. Realizando el integral obtenemos
Por lo tanto, se obtiene,
De la termodinámica estadística, sabemos que
 ,
,
de modo que sólo tenemos que diferenciar los términos que contienen V. Obtenemos
Otros parámetros termodinámicos
Reiteramos que el volumen extenso V se relaciona con el volumen por partícula v=V/N donde N = nNA es el número de partículas en el sistema. La ecuación de estado no nos da todos los parámetros termodinámicos del sistema. Podemos tomar la ecuación de la energía Helmholtz A
A partir de la ecuación deducida anteriormente para lnQ, nos encontramos con
Esta ecuación expresa A en términos de su variables naturales V y T , y por lo tanto nos da toda la información sobre el sistema termodinámico. La ecuación mecánica de estado ya se deriva por encima
La ecuación de la entropía del estado de los rendimientos de la entropía (S )
de la cual se puede calcular la energía interna
Se pueden escribir ecuaciones similares para los otros potenciales termodinámicos y químicos, pero expresando cualquier potencial en función de la presión p requerirá la solución de un polinomio de tercer orden, que produce una expresión complicada. Por lo tanto, va a ser complicado expresar la entalpía y la energía libre de Gibbs en función de sus variables naturales.
Forma reducida
A pesar de las constantes de material a y b en la forma usual de la ecuación de Van der Waals es diferente para cada líquido de una sola cuenta, la ecuación puede ser refundida en una forma invariante aplicable a todos los líquidos.
Definir las variables de reducción siguientes (
 ,
,  es la versión de reducción y variables críticas de
es la versión de reducción y variables críticas de  , respectivamente),
, respectivamente), ,
,
donde
 como muestra Salzman.[1]
como muestra Salzman.[1]
La primera forma de la ecuación de Van der Waals, mostrada más arriba, puede ser una refundición de la siguiente forma reducida:
Esta ecuación es invariante para todos los líquidos, es decir, se aplica la misma ecuación en forma reducida de estado, no importa a cual a y b puede ser que el fluido particular.
Esta invariancia también puede ser entendida en términos del principio de estados correspondientes. Si dos fluidos tienen la misma presión, volumen y temperatura reducida, podemos decir que sus estados son correspondientes. Los estados de dos fluidos puede ser correspondientes, incluso si su presión medida, volumen y temperatura sean muy diferentes. Si los estados de los dos fluidos son correspondientes, existen en el mismo régimen de la ecuación de forma reducida del Estado. Por lo tanto, van a responder a los cambios en más o menos de la misma manera, a pesar de sus características físicas mensurables puedan diferir significativamente.
Ecuación cúbica
La ecuación de Van der Waals es una ecuación cúbica de estado. Es decir, podemos escribir la ecuación en una forma cúbica del volumen. En la formulación de la ecuación cúbica reducida es la siguiente:
A la temperatura crítica, donde TR = pR = 1 tenemos como era de esperar
 ⇔ vR = 1
⇔ vR = 1
Para TR < 1, hay 3 valores de vR. Para TR > 1, existe un valor real para vR.
Aplicación a fluidos compresibles
La ecuación también se puede utilizar como una ecuación dependiente de P, V, T para fluidos compresibles, ya que, en este caso, los cambios en el volumen específico son pequeños, y se puede escribir de la siguiente manera:
donde
- p es la presión
- V es el volumen específico
- T es la temperatura
- A, B and C son parámetros.
Regla de área igual de Maxwell
Por debajo de la temperatura crítica (T’ < 1) como se muestra, oscila una isoterma de la ecuación de Van der Waals.
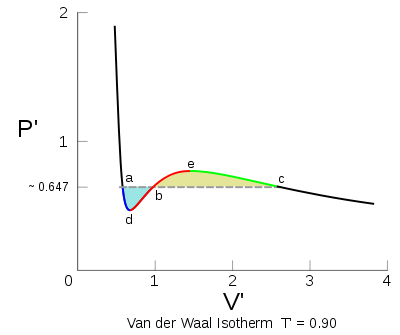 La regla de Maxwell elimina el comportamiento oscilante de la isoterma en la zona de cambio de estado definiéndolo como una cierta isobara en esa zona.
La regla de Maxwell elimina el comportamiento oscilante de la isoterma en la zona de cambio de estado definiéndolo como una cierta isobara en esa zona.A lo largo de la porción roja de la isoterma
 que es inestable, la ecuación de Van der Waals falla para describir las sustancias reales en esta región debido a que dicha ecuación siempre asume que el fluido es uniforme, mientras que entre a y c en la isoterma se vuelve más estable que un coexistencia de dos fases, una fase más densa que normalmente llamamos líquida y una fase más dispersa que normalmente llamamos gaseosa. Para solucionar este problema James Clerk Maxwell (1875) sustituyó a la isoterma de entre a y c, con una línea horizontal de modo que las áreas de las dos regiones establecidas sean iguales. La parte de la línea fija de la isoterma de ahora corresponde al equilibrio líquido-vapor. Las partes a–d y c–e se interpretan como estados metaestables de líquidos super-calientes y super-refrigerados por vapor, respectivamente.[2]
que es inestable, la ecuación de Van der Waals falla para describir las sustancias reales en esta región debido a que dicha ecuación siempre asume que el fluido es uniforme, mientras que entre a y c en la isoterma se vuelve más estable que un coexistencia de dos fases, una fase más densa que normalmente llamamos líquida y una fase más dispersa que normalmente llamamos gaseosa. Para solucionar este problema James Clerk Maxwell (1875) sustituyó a la isoterma de entre a y c, con una línea horizontal de modo que las áreas de las dos regiones establecidas sean iguales. La parte de la línea fija de la isoterma de ahora corresponde al equilibrio líquido-vapor. Las partes a–d y c–e se interpretan como estados metaestables de líquidos super-calientes y super-refrigerados por vapor, respectivamente.[2]Maxwell justifica la regla, diciendo que el trabajo realizado en el sistema al pasar desde c a b debe ser igual a trabajo liberado al pasar de a a B. (El área en el diagrama PV se corresponde a un trabajo mecánico). Eso es porque el cambio en la función energía libre A (T, V) es igual al trabajo realizado durante un proceso reversible de la función energía libre siendo una variable de estado debe tomar un valor único sin tener en cuenta de la ruta. En particular, el valor de A en el punto b debe calcular el mismo, independientemente de si la ruta procede de la izquierda o de la derecha, o se dirigen directamente a través de la isoterma horizontal o alrededor de la isoterma original de Van der Waals. El Argumento de Maxwell no es totalmente convincente, ya que requiere de un proceso reversible a través de una región de inestabilidad termodinámica. Sin embargo, los argumentos más sutiles sobre la base de las modernas teorías de equilibrio de fases parecen confirmar la construcción del área igualitaria de Maxwell y sigue siendo válida una modificación de la ecuación de Van der Waals.[3]
La regla de igual área de Maxwell se pueden derivar del supuesto de la igualdada del potencial químico, μ, en la coexistencia de las fase líquida y de vapor.[4]
Obtención de parámetros a partir del punto crítico
Los parámetros de la ecuación de Van der Waals,
 y
y  son obtenidos en general a partir de las condiciones de punto crítico de un compuesto, es decir, el punto en que las fases en equilibrio líquida y gaseosa se hacen idénticas. En dicho punto se verifica
son obtenidos en general a partir de las condiciones de punto crítico de un compuesto, es decir, el punto en que las fases en equilibrio líquida y gaseosa se hacen idénticas. En dicho punto se verifica
Donde el subíndice 'c' implica que la propiedad es la correspondiente al punto crítico. Aplicando estas condiciones a la ecuación de Van der Waals se obtienen un sistema de 3 ecuaciones, por lo que una de las propiedades deberá variar en orden de tener un sistema de ecuaciones definido. Si bien posible usar como parámetro ajustable la constante
 [cita requerida], la misma suele dejarse fija, obteniendo el límite del gas ideal en el caso de que y sean cero. El parámetro que se permite variar es entonces el volumen crítico.[cita requerida]
[cita requerida], la misma suele dejarse fija, obteniendo el límite del gas ideal en el caso de que y sean cero. El parámetro que se permite variar es entonces el volumen crítico.[cita requerida]La resolución del sistema entonces conlleva a las siguientes expresiones:



Es aquí donde se ve que fijando temperatura y presión en su punto crítico, la ecuación de Van der Waals no da buenas predicciones de los volúmenes molares de compuestos puros. Siendo el factor de compresibilidad:

Para la ecuación de Van der Waals se obtiene el valor fijo de

Como esta ecuación está derivada para moléculas con simetría esférica, si se compara este valor con el de compuestos como los gases nobles, o incluso metano, oxígeno y nitrógeno moleculares (que no son exactamente esféricos, pero se les aproximan), se encuentra que su valor experimental se haya alrededor de 0,29 aproximadamente.[5] Es entonces que la ecuación de Van der Waals original, en general, predice valores de volúmenes molares mayores (o equivalentemente, densidades menores) a las experimentales.
Enlaces externos
Véase también
- Ecuación de estado
- Ley de los gases
- Ley de los gases ideales
- Gas ideal
- Constantes de Van der Waals
- Teorema de los estados correspondientes
- Inversión de temperatura
- Construcción de Maxwell
- Iteración Para resolver ecuaciones implícitas como la de Van der Waals
Referencias
- ↑ Salzman WR, constantes crítica de los gases de Van der Waals, http://www.chem.arizona.edu/~salzmanr/480a/480ants/vdwcrit/vdwcrit.html
- ↑ Maxwell, JC Los trabajos científicos de James Clerk Maxwell, Dover 1965 (c1890) P424
- ↑ Cruz, Michael Transiciones de Fase de Primer Orden, http://www.pma.caltech.edu/~mcc/Ph127/b/Lecture3.pdf
- ↑ Elhassan AE, RJB Craven, KM de Reuck El método del área de fluidos puros y un análisis de la región de dos fases, equilibrio líquido de fases 130 (1997) 167-187.
- ↑ J.M. Smith, H.C. Van Ness, M. M. Abbott. Introducción a la Termodinámica en Ingeniería Química. 5ª Ed. McGRAW-HILL (1997). pp 726-727
Categorías:- Leyes de los gases
- Gases
- Termodinámica
- Ecuaciones
- Ecuaciones de dinámica de fluidos
- Ingeniería química

















![A(T,V,N)=N k T \left[ \ln \left(\frac{\Lambda^3 N}{(V-Nb')} \right)-1 \right] -\frac{a' N^2}{V}.](7/3375b7cc48b6d614a2eff78a968e36e2.png)

![S = -\left(\frac{\partial A}{\partial T}\right)_V
=Nk\left[ \ln\left(\frac{\Lambda^3 N}{(V-Nb')}\right)+\frac{5}{2} \right]](e/a4ed0e0672f58eef4895a1a9beb6bb02.png)





Wikimedia foundation. 2010.