- Micosis fungoide
-
Micosis fungoide 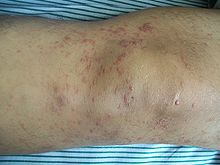
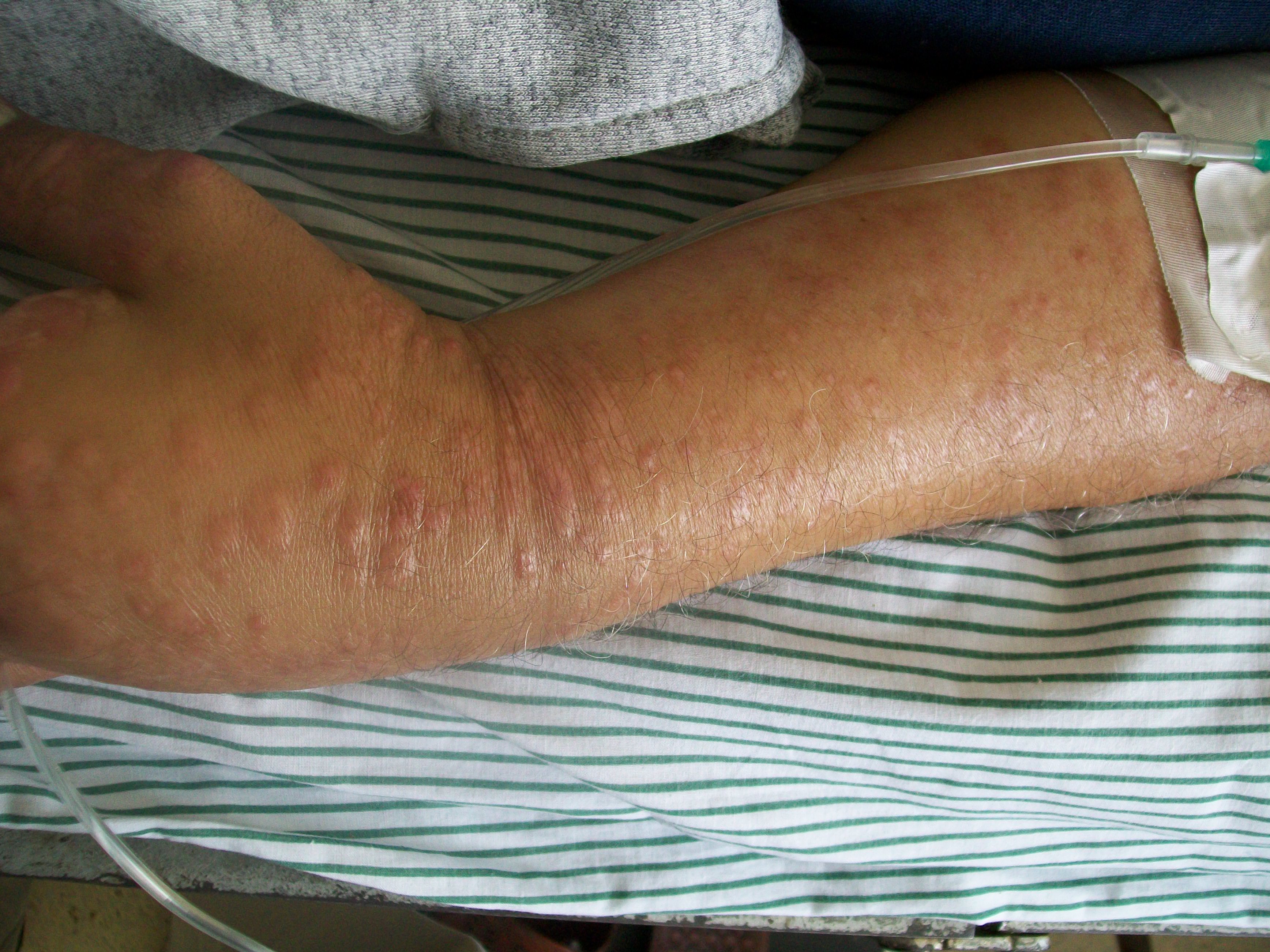
Lesiones de piel en la rodilla de un paciente de 52 años con micosis fungoideClasificación y recursos externos CIE-10 C84.0 CIE-9 202.1 OMIM 254400 DiseasesDB 8595 PubMed Buscar en Medline mediante PubMed (en inglés) eMedicine med/1541 MeSH D009182  Aviso médico
Aviso médico La micosis fungoide es el término que recibe un trastorno progresivo, crónico y no contagioso de la piel que forma parte de los linfomas no-Hodgkin, caracterizado por una proliferación de células T a nivel cutáneo.[1] La micosis fungoide es la expresión más frecuente del linfoma cutáneo de células T y se presenta como una afectación de la piel en la que aparecen brotes de lesiones planas, en forma de placas delgadas, o pequeños tumores.[2] La micosis fungoide está muy relacionada con el Síndrome de Sézary, que es una forma más agresiva del linfoma cutáneo de células T en el que la piel se ve afectada de manera difusa con considerable asociación a nivel de la sangre periférica.[3] A menudo las lesiones pueden diseminarse a los ganglios linfáticos y otros órganos.[4]
Contenido
Historia
La micosis fungoide fue descrita por primera vez en 1806 por el dermatólogo francés Jean-Louis-Marc Alibert. El nombre de la patología es un tanto engañosa, pues el término vagamente significa «enfermedad fúngica parecida a hongos» y se emplea para describir la apariencia de las lesiones y no su causa.[2] La enfermedad, sin embargo, no es una infección fúngica, sino más bien un tipo de linfoma no-Hodgkin. Fue llamada así porque Alibert describió los tumores en piel de un caso grave en un paciente de nombre Lucas de 56 años de edad como etiología fúngica, debido a su aspecto, por lo que lo describió como:
"un proceso descamativo en la piel y poco después le aparecen tumores por distintas zonas del cuerpo (…) se parecen a hongos, de consistencia como setas".[5]En 1870, Pierre-Antoine-Ernest Bazin describe con mejor detalla la micosis fungoide y divide el curso de la enfermedad.[6] Posteriormente, a finales del Siglo XIX se descubre que pueden aparecer tumores sin que éstos sean precedidos por las etapas anteriormente descritas, condición que se denominó con el término micosis fungoide demblée o «micosis fungoide primitiva».[7]
En 1979, un seminario internacional patrocinado por el Instituto Nacional del Cáncer estadounidense acuñó el término linfoma cutáneo de células T (LCCT) y se emplea en el presente para describir un grupo heterogéneo de linfomas primarios compuestas por células T malignas con manifestaciones en la piel, como la micosis fungoide.[8]
Etiología
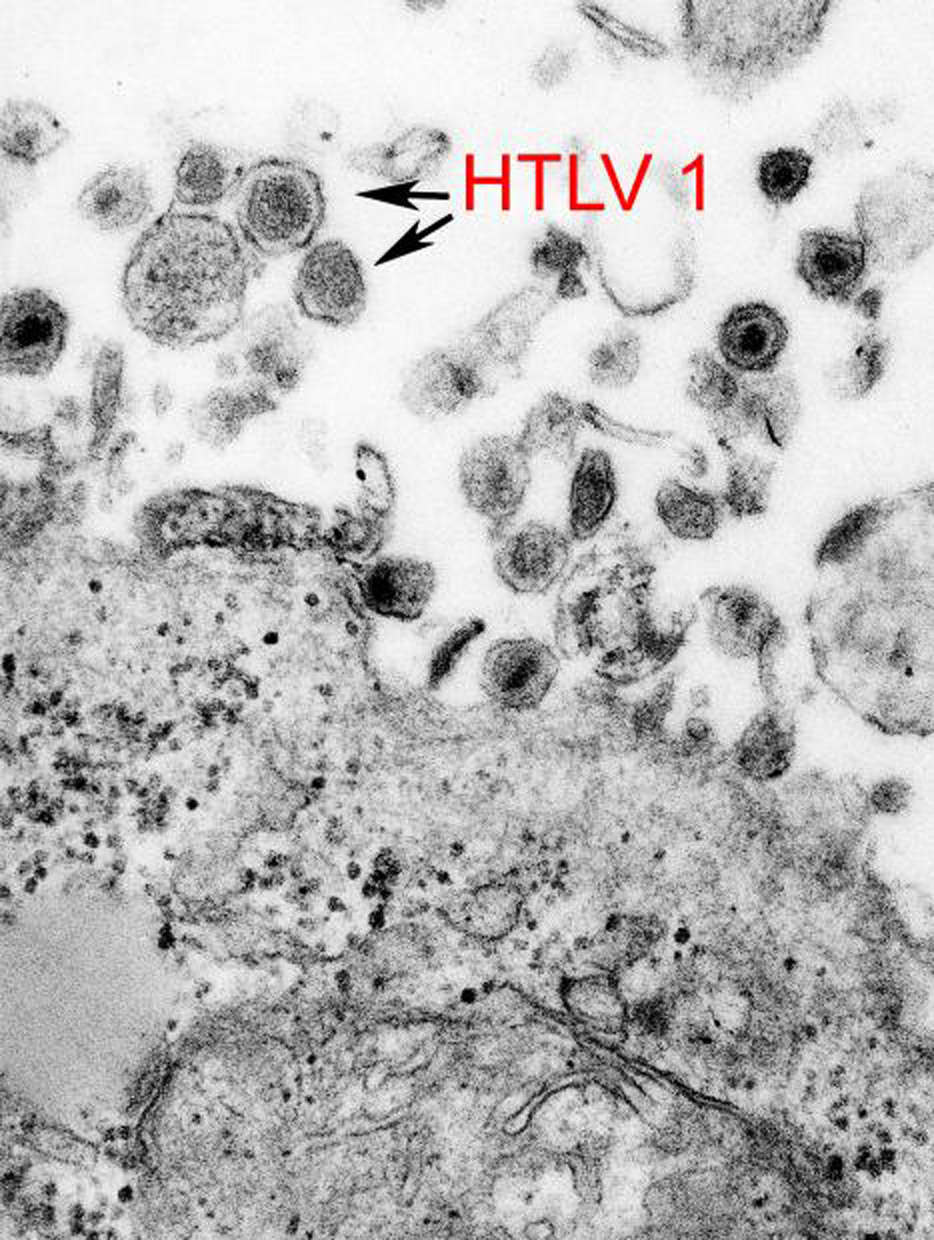
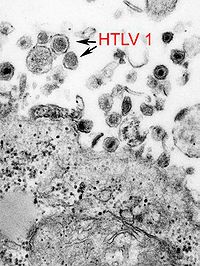 El Virus 1 Linfotrópico T Humano (VLTH-1) está asociado a neoplasia.s de las líneas de linfocitos T, y por lo tanto, probablemente también a la etiología de la micosis fungoide.[9]
El Virus 1 Linfotrópico T Humano (VLTH-1) está asociado a neoplasia.s de las líneas de linfocitos T, y por lo tanto, probablemente también a la etiología de la micosis fungoide.[9]
La causa específica de la micosis fungoide permanece aún desconocida, aunque, en la gran mayoría de los casos no se cree que sea de causa genética o hereditaria. Se ha informado de un caso con posible vínculo genético. Se ha sospechado como una de las posibles causas la infección al retrovirus VLTH-1.[4] Hasta ahora no existen evidencias convincentes que demuestren una asociación entre la exposición a sustancias químicas y los linfomas cutáneos de células T, incluyendo la micosis fungoide.[2]
No es frecuente que la enfermedad aparezca antes de los 20 años de edad, y parece ser notablemente más frecuente en varones que en mujeres,[4] especialmente mayores de 50 años, donde la incidencia de la enfermedad aumenta. La edad media de aparición es de entre 45 y 55 años de edad para los pacientes con la variante de la enfermedad que solo produce lesiones en piel tipo parches y placas, pero la edad media sube a más de 60 años para pacientes que se presentan con tumores, enrojecimiento de la piel o en su forma leucémica, el síndrome de Sezary.
Patogenia
La enfermedad se caracteriza por una inusual expresión de células T CD4, parte del sistema inmunitario. Estas células T están clásicamente asociadas a la piel, lo que significa que bioquímicamente y biológicamente se relacionan más dinamicamente con la piel que otros órganos. La micosis fungoide es el tipo más común de linfoma cutáneo de células T (LCCT), pero hay muchos otros tipos de LCCT que no tienen nada que ver con la micosis fungoide y estos trastornos son manejados de manera diferente.
Se ha demostrado la presencia de rearreglo genético por medio de técnicas moleculares y citogenéticas y/o de expansión de células T con inmunofenotipo de células T malignas, en particular un aumento en las poblaciones de células CD4+ de tal manera que la relación CD4/CD8 resulta >10 y/o una expansión en la población de células T con pérdida de uno o más de sus antígenos comunes de superficie como los cúmulos de diferenciación CD2, CD3 o CD5. Las células T malignas circulantes suelen tener los marcadores CD7 y CD26-.[8]
Cuadro clínico
Los síntomas típicos visibles incluyen lesiones tipo máculas o pápulas que no suelen ulcerar y pueden ser descamativas. En aproximadamente 20% de los pacientes, las lesiones causan picor. La enfermedad cutánea puede persisitir por varios años, especialmente en pacientes que se automedican. Por lo general se ven afectados los ganglios linfáticos del sujeto, lo cual se revela como un aumento de volumen a nivel del cuello por debajo de la mandíbula o en la axila.
Estadificación
El curso clínico de la micosis fungoide avanza en tres etapas.[10] La primera etapa es premicótica o eritematosa, caracterizado por erupción localizada que suele causar picor muy parecido a las lesiones de la psoriasis[5] o una dermatitis atópica.[2] La segunda etapa se caracteriza por la aparición de manchas en forma de placas induradas, con bordes bien delimitados y ligeramente elevadas por encima del plano de la piel.[10] Finalmente, en la tercera etapa aparecen lesiones tipo tumor o placas protuberantes y que ocasionalmente pueden volverse violáceas y formar pequeñas úlceras.[5]
Clásicamente se divide a la micosis fungoide en 5 estadios:
Estadio Características de la lesión eritematosa[11] I Mínima (<10%) infiltración focal de la piel II Muestra infiltración cutánea moderada sin infiltración ganglionar III Afectación cutánea generalizada con pariencia en placas o nodular IV A las lesiones generalizadas de la pies se asocia metástasis a ganglios linfáticos V Afectación visceral y en sangre periférica además de las lesiones en piel y ganglios linfáticos Diagnóstico
 La ausencia en la expresión del cúmulo de diferenciación (CD) CD26 sobre linfocitos T es un marcador diagnóstico de un linfoma de células T en sangre periférica.[12]
La ausencia en la expresión del cúmulo de diferenciación (CD) CD26 sobre linfocitos T es un marcador diagnóstico de un linfoma de células T en sangre periférica.[12]El diagnóstico es a veces difícil porque las primeras fases de la enfermedad suelen ser semejantes al eccema, o incluso la psoriasis. Como con cualquier enfermedad grave, es aconsejable seguir la opinión de un profesional médico si se sospecha de un caso de micosis fungoide. El diagnóstico es generalmente realizado a través de una biopsia de la piel. Para que el diagnóstico sea más certero, a menudo se recomienda la toma de más de una biopsia. El diagnóstico se realiza a través de una combinación del cuadro clínico y el examen físico, y se confirma por los resultados de la biopsia.
Para la estadificación del trastorno, se suelen solicitar varias pruebas paraclínicas, incluyendo una evaluación médica de los ganglios linfáticos, pruebas bioquímicas en la sangre y en órganos internos afectados, aunque la mayoría de los pacientes presentan enfermedad aparentemente limitada a la piel, en forma de manchas planas enrojecidas o violáceas y en forma de placas o ligeramente elevados y «arrugados».
Tratamiento
La micosis fungoide puede ser tratada de diversas maneras. Los tratamientos más comunes incluyen la simple luz solar, la luz ultravioleta, esteroides tópicos y sistémicos, quimioterapia, radioterapia superficial localizado, el vorinostat que es un inhibidor de la histona deacetilasa, la radioterapia corporal total con haz de electrones sobre la piel, y terapias biológicas como los interferones, los retinoides—que son análogos de la vitamina A—, y rexinoides.[2] Los tratamientos a menudo se utilizan en combinación.
La selección de tratamientos por lo general depende de la preferencia del paciente y el acceso a dichas terapias, así como las recomendaciones de los médicos, la etapa de la enfermedad, pacientes con antecedentes de resistencia a terapias en el pasado, las alergias del paciente, evidencia clínica de un beneficio positivo: proporción de riesgo, y así sucesivamente.
Es discutible si la cura fue realmente obtenida a través de algún tipo específico de tratamientos, pues muchos pacientes experimentan períodos prolongados de control de la enfermedad y al menos la mitad de los pacientes no mueren de esta enfermedad, incluso si no son realmente curados.
La calidad de vida es uno de los principales objetivos, además que el curar, y maximizar los períodos de remisión o de estabilización de la enfermedad, así como reducir al mínimo los efectos del tratamientos y la toxicidad, son dos preocupaciones centrales en la atención clínica.
Pronóstico
Si el tratamiento tiene éxito la enfermedad puede entrar en un estado que deja de progresar con un examen clínico libre de patologías y diversas pruebas con resultados negativos. Esto se conoce como el estado de remisión, que puede durar indefinidamente.
Los tratamientos también pueden causar enfermedad deje de avanzar, mientras que sigue presente en el sujeto, y a esto se le conoce como un estado de enfermedad estable, que también puede durar indefinidamente, pero resulta ser una situación más grave. La enfermedad también puede avanzar, con asociación de ganglios linfáticos, la sangre y órganos internos, o se transforma en un grado de linfoma más avanzado.
El síndrome de Sezary es una variante de la micosis fungoide, que aparece en alrededor de 5% de todos los casos de micosis fungoide.[8] El paciente con síndrome de Sezary cursa con eritrodermia exfoliativa generalizada, linfadenopatía, y más de 1000 linfocitos atípicos por mm3 de sangre periférica con marcadores para linfocitos T y núcleos cerebriformes, o bien son positivos para otras pruebas que confirmen la presencia de líneas malignsa de células T en la sangre, incluyendo la reordenación de genes en las células clonales de células T del mismo patrón que las células T que se encuentra en la piel.
Referencias
- ↑ Instituto Nacional del Cáncer. «Información general sobre la micosis fungoide y el síndrome de Sézary» (en español). Consultado el 27 de junio de 2009.
- ↑ a b c d e Rigel, Darrell S; Robert Friedman, Leonard M. Dzubow, Douglas S. Reintgen, M.D., Jean-Claude Bystryn y Robin Marks (2006) (en español). Cáncer de piel. Elsevier, España. pp. 349-358. ISBN 8481748757. http://books.google.es/books?id=E2XdJA8AbqQC&source=gbs_navlinks_s.
- ↑ Girardi, M; Heald PW, Wilson LD. (mayo 2004). «The pathogenesis of mycosis fungoides.». N Engl J Med. May 350 (19): pp. 1978-88. PMID 15128898. http://content.nejm.org/cgi/content/extract/350/19/1978. Consultado el 27 de junio de 2009.
- ↑ a b c Ferri, Fred F. (2006). Ferri consultor clínico, 2006-2007: Claves diagnósticas y tratamiento. Elsevier, España. pp. 550. ISBN 8481749141. http://books.google.es/books?id=pMQfIaasmV0C&source=gbs_navlinks_s.
- ↑ a b c d GARZONA NAVAS, Dra. Laura, MOREIRA HIDALGO, Dr. Federico, HIDALGO MATLOCK, Dr. Benjamín et al. Micosis Fungoide: Revisión de tema y presentación de un caso. Rev. costarric. salud pública. [online]. jul. 2007, vol.16, no.30 [citado 27 Junio 2009], p.46-53. Disponible en la World Wide Web: [1]. ISSN 1409-1429.
- ↑ Du Vivier, Anthony; Phillip H. McKee (2002) (en inglés). Atlas of clinical dermatology (3ra edición). Elsevier Health Sciences. ISBN 0443072205. http://books.google.es/books?id=LxoiVpwpWdMC&source=gbs_navlinks_s.
- ↑ Revista de medicina y cirugia practicas (en español). Publicado en 1911, pág 335, 405-406. Procedencia del original: la Universidad de California. Digitalizado 16 Abr 2007.
- ↑ a b c Pinter-Brown, Lauren C (octubre de 2008). «Mycosis fungoides» (en inglés). Stem Cells and Disorders. eMedicine.com. Consultado el 28 de junio de 2009.
- ↑ Kumar, Vinay; Cotran, Ramzi S.; Robbins Stanley L. (2003). «Lesión, adaptación y muerte celular». Patología humana: Robbins (Séptima edición). Elsevier España. ISBN 8481746665. «Véase página 20 y 616.»
- ↑ a b Laskaris, George (2005). Atlas de enfermedades orales. Elsevier, España. pp. 362. ISBN 8445813293. http://books.google.es/books?id=zwOpLEQc2eIC&source=gbs_navlinks_s.
- ↑ National Cancer Institute. «Stages of Mycosis Fungoides and the Sézary Syndrome» (en inglés). Consultado el 28 de junio de 2009.
- ↑ Jones, D; Dang NH, Duvic M, Washington LT, Huh YO. (mayo 2004). «Absence of CD26 expression is a useful marker for diagnosis of T-cell lymphoma in peripheral blood.». Am J Clin Pathol. 115 (6): pp. 885-92. PMID 11392886. http://ajcp.ascpjournals.org/content/115/6/885.long. Consultado el 27 de junio de 2009.
Categorías:- Sistema linfático
- Cáncer de piel
- Enfermedades hematológicas
Wikimedia foundation. 2010.