- Relación cuantitativa estructura actividad
-
Relación cuantitativa estructura actividad
{La relación cuantitativa estructura-actividad (en inglés, Quantitative structure-activity relationship, QSAR, o bien, quantitative structure-property relationship, QSPR, es el proceso por el cual la estructura química se correlaciona cuantitativamente con un proceso bien definido, como la actividad biológica (unión de un fármaco con un receptor) o la reactividad química (afinidad de una sustancia por otra para que produzcan una reacción).
Por ejemplo, la actividad biológica se puede expresar cuantitativamente como la concentración requerida de una substancia para dar una cierta respuesta biológica. Además, cuando las propiedades fisicoquímicas o las estructuras se expresan mediante números, podemos construir una relación matemática, o relación cuantitativa estructura-actividad, entre las dos. La expresión matemática puede entonces usarse para predecir la respuesta de otras estructuras químicas.
La forma matemática más general de QSAR es:
Este método asigna parámetros a cada grupo químico, de forma que al modificar la estructura química puede valorarse la contribución de cada grupo funcional a la actividad del fármaco o del tóxico en cuestión y a partir de ahí, cómo variará la actividad de esa sustancia.[1]
Contenido
Antecedentes históricos
Los primeros acercamientos a la correlación entre la estructura de las moléculas y sus propiedades o actividades podemos encontralos en:
- Cros estudió en 1863 el aumento del punto de ebullición y del punto de fusión, en alcanos, al aumentar el número de átomos de Carbono y la masa molecular. Asimismo observó una disminución de la solubilidad en agua, en alcoholes, al aumentar el número de átomos de Carbono y la masa molecular.[2]
- Hans Horst Meyer en 1899 y Charles Ernest Overton en 1901 observan un aumento en la toxicidad de los alcoholes al aumentar logP (coeficiente de partición aceite/agua),lo que resulta un caso típico de relación entre propiedades fisicoquímicas y actividad biológica.
- Corwin Hansch y su equipo, en 1964, promovieron la hipótesis QSAR ("moléculas semejantes tendrán un comportamiento semejante"), que cuantificaron en forma de ecuación:
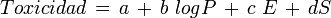
logP: coeficiente de reparto = término que cuantifica las mayores o memores cualidades hidrófobas.
E: término que cuantifica los aspectos electrónicos de la molécula.
S: término que cuantifica los aspectos estéricos y de forma.
El primero de esos téminos señala la probabilidad o capacidad de alcanzar la molécula-diana. Los otros dos términos señalan la probabilidad de interaccionar de modo eficaz con esa diana.
La relación estructura-actividad (SAR) y la paradoja de la relación estructura-actividad (SAR)
La suposición básica de todas las hipótesis basadas en la molécula es que moléculas semejantes tendrán una actividad similar. Este principio cualitativo es también llamado relación estructura-actividad o SAR, Structure-Activity Relationship. El problema subyacente es cómo definir una pequeña diferencia a nivel molecular, porque cada tipo de actividad (por ejemplo, capacidad de reacción química, capacidad de biotransformación, solubilidad, etc) podría depender de otra diferencia. Un buen ejemplo fue dado con la revisión del bioisosterismo de Patanie/LaVoie.[3]
En general,estamos más interesados en encontrar tendencias claras. Las hipótesis dependen usualmente de un conjunto finito de datos químicos.
Luego, el principio de inducción debería ser respetado para evitar un exceso de hipótesis y derivar hipótesis en exceso y derivar hipótesis excedentes e interpretaciones inútiles sobre los datos estructurales/moleculares.
La paradoja del SAR se refiere al hecho de que no es cierto que todas las moléculas similares tengan actividades similares. [4]
Tipos
Basada en el fragmento (contribución del grupo)
Se ha demostrado que logP (logaritmo del coeficiente de reparto) de un compuesto se puede determinar por la suma de los fragmentos que componen su molécula. Los valores de logP fragmentarios se han determinado estadísticamente. Este método da resultados mixtos y generalmente no se confía en que proporcione una precisión de ±0.1 unidades.[5]
3D-QSAR
3D-QSAR se refiere a la aplicación de cálculos del campo de fuerza que necesitan estructuras tridimensionales, como la cristalografía de proteínas o la superposición de moléculas. Emplea potenciales calculados, por ejemplo el potencial de Lennard-Jones, antes que constantes experimentales y se interesa por la molécula completa antes que por un simple sustituyente. Examina los campos estéricos (forma de la molécula) y los campos electrostáticos basados en la función de energía aplicada.[6]
El espacio de datos creado habitualmente se reduce posteriormente siguiendo una extracción de variables (ver también reducción de dimensionalidad). El siguiente método de aprendizaje puede ser cualquiera de los ya mencionados métodos de máquinas de aprendizaje, como las máquinas de vectores de soporte.[7]
La mayoría de los estudios indican que los químicos prefieren los métodos de mínimos cuadrados parciales (PLS o Partial Least Squares), ya que aplican extracción de variables y el la inducción en un solo paso.
Minería de datos
Para el proceso de codificación es necesario habitualmente calcular un gran número de variables o descriptores moleculares, que pueden carecer de la capacidad de interpretación estructural. En combinación con los últimos métodos de aprendizaje aplicado o durante la etapa de preprocesamiento, se produce un problema de selección de variables
Algunos ejemplos típicos de predicción basados en la minería de datos son: las máquinas de vectores de soportes, los árboles de decisión y las redes neurales para inducir un modelo predictivo de aprendizaje.
Las propuestas de minería de moléculas, un caso especial de minería de datos estructurados, aplica una matriz de similaridad (matriz de adyacencia o matriz de distancia), o bien, un esquema de fragmentación automática en subestructutas moleculares. Existen además otras propuestas que emplean el grafo molecular, una representación de los enlaces de la molécula, que a su vez, puede ser subdividido en subgrafos que dependen del número de ejes interconectados al vértice. Estos subgrafos se clasifican según su orden y su tipo y se puede encontrar semejanza entre moléculas tras la búsqueda del máximo subgrafo común o gráfico kernel.[8] [9]
Parámetros o descriptores moleculares
La elección de variables que se realiza en un esquema 3D-QSAR conduce a un conjunto de parámetros o descriptores moleculares, que se pueden calcular a partir de datos experimentales o de modelos teóricos (Método de Hansch/Fujita, Método de Nys y Rekker, Método ΣF-SYBYL de Rekker, CLogP, MOLCAD, Chemical-2) .
Entre estos parámetros podemos encontrar:
- Descriptores de propiedades fisicoquímicas:
- Descriptores de efectos hidrófobos: Como el coeficiente de reparto o de partición aceite/agua, una razón de las concentraciones de una sustancia entre dos fases en equilibrio, o la constante hidrofóbica del sustituyente, π.
- Descriptores de efectos electrónicos: Estos efectos son el efecto inductivo (F) y la resonancia (R) y son estudiados mediante la ecuación de Hammett.
- Descriptores de efectos estéricos: Se emplea el parámetro Es, factor estérico de Tafts, con el coeficiente δ que representa la sensibilidad de la reacción a los efectos estéricos, los parámetros de Verloop y Tipker o la refractividad molar.
- Descriptores de propiedades topológicas
- índices de conectividad
- Descriptores de propiedades estructurales
- presencia de subestructuras o grupos funcionales (sustituyentes en orto de un centro de reacción, sustituyentes contiguos o grupos formadores de puentes de hidrógeno)
- frecuencia en presencia
- Descriptores de propiedades electrónicas: constante de ionización y momento dipolar.
- a partir de cálculos de orbitales
- Descriptores de propiedades geométricas
- a partir de cálculos de superficies
Juzgando la calidad de los modelos QSAR
La relaciones QSAR representan modelos estadísticos predictivos derivados de la aplicación de herramientas estadísticas que correlacionan la actividad biológica (incluyendo el efecto terapéutico deseable y los efectos secundarios no deseables) de las sustancias químicas (drogas/tóxicos/contaminantes) con descriptores representativos de la estructura molecular y/o propiedades moleculares. QSAR se está aplicando en muchas disciplinas como evaluación de riesgos, predicción de toxicidad, y decisiones regulatorias[10] además de descubrimiento de fármacos y optimización de productos.[11] Obtener un modelo QSAR de buena calidad depende de muchos factores,tales como la calidad de los datos biológicos, la elección de descriptores y métodos estadísticos. Cualquier modelización QSAR debería conducir en último término a modelos estadísticamente robustos capaces de hacer predicciones precisas y fiables de las actividades biológicas de los nuevos compuestos.
Para validar los modelos QSAR se adoptan habitualmente cuatro estrategias: [12]
- validación interna o validación cruzada (validación cruzada)[13] ;
- validación por división del conjunto de datos en compuestos (muestras) de entrenamiento y de prueba;
- validación externa verdadera por aplicación del modelo sobre datos externos y
- aleatorización de datos o Y-scrambling.
El éxito de cualquier modelo QSAR depende de la precisión de los datos iniciales, selección de los descriptores apropiados y herramientas estadisticas, y lo más importante,la validación del modelo desarrollado. La validacion es el proceso por el cual se establecen la fiabilidad y relevancia de un procedimiento para un propósito es.[14] Dejar algo fuera de la validación cruzada conduce generalmente a una sobrestimación de la capacidad predictiva e, incluso con validación externa, no podemos asegurar si la selección de los conjuntos de muestras de entrenamiento y de prueba fueron manipulados para maximizar la capacidad predictiva del modelo publicado. Entre los aspectos de la validación de los modelos QSAR que deben revisarse cuidadosamente se incluyen los métodos de selección de los compuestos del conjunto de entrenamiento,[15] el modo de establecer el tamaño del conjunto de entrenamiento [16] y el impacto de la selección de variables [17] sobre los modelos del conjunto de entrenamiento para determinar la calidad de la predicción. Ee también importante el desarrollo de nuevos parámetros de validación para juzgar la calidad de los modelos QSAR .[18]
Aplicaciones
Químicas
Una de las primeras aplicaciones históricas de las aplicaciones QSAR fue la predicción de los puntos de ebullición. [19]
Es bien conocido, por ejemplo, que dentro de una determinada familia de compuestos químicos, especialmente de química orgánica, existe una correlación fuerte entre la estructura y las propiedades observadas. Un ejemplo sencillo es la relación entre el número de átomos de carbono en alcanos y su punto de ebullición. Hay una tendencia clara en elaumento del punto de ebullición con el incremento del número de átomos de carbono y esto sirve como medio de predecir el punto de ebullición de los alcanos superiores.
Otras aplicaciones muy interesantes son los métodos de la ecuación de Hammett, que se indica más abajo, la ecuación de Taft y la predicción de la constante de disociación de los ácidos, Ka.[20]
En la ecuación de Hammett anterior, se relacionan las constantes de dissociación de dos reacciones de ionización del àcido benzoico con el anillo benzoico sin sustituir KH) y del ácido fenilacético con elanillo también sin sustituir, (K'H). El valor σ puede tomar tres tipos de valores numéricos según el tipo de sustituyente: σ=0 cuando el sustituyente es el hidrógeno, σ>0 cuando es un aceptor de electrones y σ<0 cuando es un donador de electrones. El valor de ρ (pendiente de la función de la ecuación) mide la sensibilidad de la reacción: el valor puede ser positivo o negativo,; si ρ es grande, la reacción es muy sensible; y el signo del valor refleja la presencia de cargas negativas o positivas. No obstante, existen tres excepciones a la función lineal de la ecuación de Hammett: p-CO2Et, p-SO2Me, p-NO2, debido a la resonancia. En estos casos se introducen nuevos parámetros:
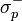 (para sustituyentes aceptores de electrones que interactúan por resonancia con un centro de reacción rico en electrones) y
(para sustituyentes aceptores de electrones que interactúan por resonancia con un centro de reacción rico en electrones) y 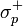 (para un grupo donador que por resonancia interactúa con un centro de reacción deficiente en electrones).
(para un grupo donador que por resonancia interactúa con un centro de reacción deficiente en electrones).Biológicas
La actividad biológica de las moléculas se mide habitualmente en ensayos para establecer el nivel de inhibición de determinada transducción de señal o ruta metabólica. Las sustancias químicas pueden ser también biológicamente activas si poseen toxicidad. El descubrimiento de fármacos, a menudo, supone el uso de QSAR para idebtificar las estructuras químicas que podrían tener buenos efectos inhibidores sobre una diana biológica específica y tener baja toxicidad (actividad no-específica). La predicción del coeficiente de partición, log P, es de especial interés. Es una medida importante en la identificación de sustancias con potencial farmacológico de acuerdo con la Regla de cinco de Lipinski.
Mientras que muchos análisis de las relaciones cuantitativas estructura-actividad suponen la interacción de una familia de moléculas con un enzima o receptor uniéndose al sitio, QSAR puede usarse también para estudiar las interacciones en el dominio estructural de las proteínas. Las interacciones proteína-proteína pueden analizarse cuantitativamente mediante las variaciones estructurales resultado de la mutagénesis dirigida por el sitio.[21]
Eso es parte del método del aprendizaje automático que reduce el riesgo de la paradoja SAR (relación estructura-actividad), especialmente teniendo en cuenta que sólo está disponible una cantidad finita de datos. (Véase también el Estimador No-sesgado de mínima varianza, MVUE). En general todos los problemas QSAR pueden dividirse en codificación[22] y aprendizaje.[23]
Dominio de aplicabilidad
Como el uso de los modelos (Q)SAR para gestión del riesgo químico aumenta de modo constante y también se emplea para propósitos de regulación, es de crucial importancia que sea capaz de evaluar la fiabilidad de las predicciones. El espacio de descriptores químicos extendido para un determinado conjunto de entrenamiento de compuestos químicos se llama dominio de aplicabilidad. Ofrece la oportunidad de evaluar si se pueden hacer predicciones fiables sobre un compuesto.
Véase también
- Quimioinformática
- ADME
- Solubilidad diferencial
- Fuerza intermolecular
- Farmacocinética
- Farmacóforo
- CLogP
- Diseño de fármacos asistido por ordenador (CADD)
- Software de diseño molecular
- Predicción de estructura de proteínas
- QSAR & Ciencia combinatoria – Revista científica
- Software para modelado de la mecánica molecular
Referencias
- ↑ Toxicología básica o fundamental. Tema 2. Antonio Hernández Jerez. Departamento de Medicina Legal, Toxicología y Psiquiatría. Facultad de Medicina. Universidad de Granada
- ↑ Correlaciones estructura-actividad. Optimización de prototipos. María Font. Dpto. Química Orgánica y Farmacéutica. Sección de Modelización Molecular. Universidad de Navarra.
- ↑ Patani GA, LaVoie EJ (December de 1996). «Bioisosterism: A Rational Approach in Drug Design» Chemical Reviews. Vol. 96. n.º 8. pp. 3147–3176. DOI 10.1021/cr950066q. PMID 11848856.
- ↑ http://blog.alquimia.com/Default.aspx?tabid=289&EntryID=83 P.S.R. Prevention Safety Regulatory.
- ↑ Wildman SA, Crippen GM (1999), "Prediction of physicochemical parameters by atomic contributions", J. Chem. Inf. Comput. Sci 39 (5): 868–873, doi:
- ↑ Leach, Andrew R. (2001). Molecular modelling: principles and applications. Englewood Cliffs, N.J: Prentice Hall. ISBN 0-582-38210-6.
- ↑ Vert, Jean-Philippe; Schl̲kopf, Bernhard; Schölkopf, Bernhard; Tsuda, Koji (2004). Kernel methods in computational biology. Cambridge, Mass: MIT Press. ISBN 0-262-19509-7.
- ↑ Gusfield, Dan (1997). Algorithms on strings, trees, and sequences: computer science and computational biology. Cambridge, UK: Cambridge University Press. ISBN 0-521-58519-8.
- ↑ Helma, Christoph (2005). Predictive toxicology. Washington, DC: Taylor & Francis. ISBN 0-8247-2397-X.
- ↑ Tong W, Hong H, Xie Q, Shi L, Fang H, Perkins R (April de 2005). «Assessing QSAR Limitations – A Regulatory Perspective» Current Computer-Aided Drug Design. Vol. 1. n.º 2. pp. 195–205.
- ↑ Dearden JC (2003). «In silico prediction of drug toxicity» Journal of Computer-aided Molecular Design. Vol. 17. n.º 2–4. pp. 119–27. DOI 10.1023/A:1025361621494. PMID 13677480.
- ↑ Wold S, Eriksson L (1995). «Statistical validation of QSAR results», Waterbeemd, Han van de (ed.). Chemometric methods in molecular design. Weinheim: VCH, pp. 309–318. ISBN 3-527-30044-9.
- ↑ Introducción al diseño racional de fármacos. J.C. Escalona, R. Carrasco y J. A. Padrón. Ciudad de La Habana : Editorial Universitaria, 2008. ISBN 978-959-16-0647-1. Página 36
- ↑ Roy, K. (2007), "On some aspects of validation of predictive quantitative structure-activity relationship models", Expert Opin. Drug Discov. 2 (12): 1567–1577, doi:
- ↑ Leonard JT, Roy K (2006), "On selection of training and test sets for the development of predictive QSAR models", QSAR & Combinatorial Science 25 (3): 235–251, doi:, http://library.sut.ac.th/free/ph_scie/pdfs/qsar/QSAR_3.pdf
- ↑ Roy PP, Leonard JT, Roy K (2008), "Exploring the impact of size of training sets for the development of predictive QSAR models", Chemometrics and Intelligent Laboratory Systems 90 (1): 31–42, doi:
- ↑ Roy PP, Roy K (2008), "On some aspects of variable selection for partial least squares regression models", QSAR & Combinatorial Science 27 (3): 302–313, doi:
- ↑ Roy PP, Paul S, Mitra I, Roy K (April de 2009). «On Two Novel Parameters for Validation of Predictive QSAR Models» Molecules. Vol. 14. n.º 5. pp. 1660–1701. DOI 10.3390/molecules14051660.
- ↑ Rouvray, D. H.; Bonchev, Danail (1991). Chemical graph theory: introduction and fundamentals. Tunbridge Wells, Kent, England: Abacus Press. ISBN 0-85626-454-7.
- ↑ Fraczkiewicz R (2007). «In Silico Prediction of Ionization», Comprehensive medicinal chemistry II. Amsterdam: Elsevier. ISBN 0-08-044518-7.
- ↑ Freyhult EK, Andersson K, Gustafsson MG (April de 2003). «Structural modeling extends QSAR analysis of antibody-lysozyme interactions to 3D-QSAR» Biophysical Journal. Vol. 84. n.º 4. pp. 2264–72. PMC 1302793. DOI 10.1016/S0006-3495(03)75032-2. PMID 12668435.
- ↑ Hendrik Timmerman; Todeschini, Roberto; Viviana Consonni; Raimund Mannhold; Hugo Kubinyi (2002). Handbook of Molecular Descriptors. Weinheim: Wiley-VCH. ISBN 3-527-29913-0.
- ↑ Strok, David G.; Duda, Richard O.; Hart, Peter W. (2001). Pattern classification. Chichester: John Wiley & Sons. ISBN 0-471-05669-3.
Lecturas adicionales
- Avendaño López, Carmen. Introducción a la Química Farmacéutica. Ed. MacGraw-Hill Interamericana. 1993.
- Selassie CD (2003). «History of Quantitative Structure-Activity Relationships», Abraham DJ (ed.). Burger's medicinal Chemistry and Drug Discovery, 6th edición (vol. 1), New York: Wiley, pp. 1–48. ISBN 0-471-27401-1.
Enlaces externos
- Correlaciones estructura-actividad. Optimización de prototipos. María Font. Dpto. Química Orgánica y Farmacéutica. Sección de Modelización Molecular. Universidad de Navarra.
- Informática biomédica y su impacto en el I+D de medicamentos. Ferran Sanz. Programa de Investigación en Informática Biomédica (GRIB). IMIM - Hospital del Mar. Universidad Pompeu Fabra. Parque de Investigación Biomédica de Barcelona (PRBB).
- Introducción al diseño racional de fármacos. J.C. Escalona, R. Carrasco y J. A. Padrón. Ciudad de La Habana : Editorial Universitaria, 2008. ISBN 978-959-16-0647-1
- «The Cheminformatics and QSAR Society». Consultado el 2009-05-11.
- «Nature Protocols: Development of QSAR models using C-QSAR program». Nature Protocols. doi:10.1038/nprot.2007.125. Consultado el 2009-05-11. «A regression program that has dual databases of over 21,000 QSAR models»
- «QSAR World». Consultado el 2009-05-11. «A comprehensive web resource for QSAR modelers»
Categorías: Química farmacéutica | Farmacología | Quimioinformática | Fisicoquímica
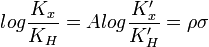
Wikimedia foundation. 2010.