- Farmacocinética
-
Farmacocinética

La farmacocinética es la rama de la farmacología que estudia los procesos a los que un fármaco es sometido a través de su paso por el organismo. Trata de dilucidar qué sucede con un fármaco desde el momento en el que es administrado hasta su total eliminación del cuerpo.
Para ello, se han desarrollado diferentes modelos que simplifiquen los numerosos procesos que tienen lugar entre el organismo y el fármaco. Aún cuando dentro de los mismos el modelo policomportamental es el más próximo a la realidad, la complicación que conlleva ha hecho que sean los modelos monocompartimental y en todo caso el bicompartimental los más usados. Desde esos prismas, el estudio detallado de los sucesivos pasos que atraviesa el fármaco en el organismo, se agrupan bajo el anagrama LADME:
- Liberación del producto activo,
- Absorción del mismo,
- Distribución por el organismo,
- Metabolismo o inactivación, al ser reconocido por el organismo como una sustancia extraña al mismo, y
- Eliminación del fármaco o los residuos que queden del mismo.
Estas distintas fases, implican la utilización y manejo de conceptos básicos para comprender la dinámica instaurada. Así, las propiedades de las sustancias que actúan como excipientes, las características de las membranas biológicas y la forma en que las sustancias pueden atravesarlas, o las características de las reacciones enzimáticas que inactivan al fármaco, son de necesario conocimiento para la correcta comprensión de la cinética del fármaco.
Todos estos conceptos se pueden representar mediante fórmulas matemáticas que tienen su correspondiente representación gráfica. De esta manera se puede conocer tanto las características de una molécula, así como la manera en que se comportará determinado fármaco conociendo algunas de sus características básicas. Así, el conocimiento del pK, su biodisponibilidad o hidrosolubilidad, orienta sobre su capacidad de absorción o distribución en el organismo.
Las gráficas resultantes del estudio de un fármaco tienen valor trascendente en aplicaciones en la industria (cálculos de bioequivalencia en el diseño de fármacos genéricos, por ejemplo) o en la aplicación clínica de los conceptos farmacocinéticos. En efecto, la farmacocinética clínica provee abundantes pautas de actuación para el correcto manejo de los fármacos, buscando el máximo de efectividad y utilidad para los profesionales de la medicina humana y veterinaria.
Contenido
Modelos farmacocinéticos
El resultado final de las transformaciones que sufre un fármaco en el organismo y las reglas que las rigen, depende de la suma de múltiples factores habitualmente interrelacionados entre sí. Con el objetivo de simplificar el estudio se diseñaron modelos de funcionamiento basados fundamentalmente en la consideración del organismo como compartimentos relacionados entre sí. Conceptualmente, la propuesta más simple es la consideración homogénea del organismo, con la existencia de un solo compartimento. Este modelo monocompartimental presupone que las concentraciones plasmáticas del fármaco son fiel reflejo de las concentraciones en otros fluidos o tejidos, y que la eliminación del fármaco es directamente proporcional a los niveles en el organismo del fármaco (cinética de primer grado).
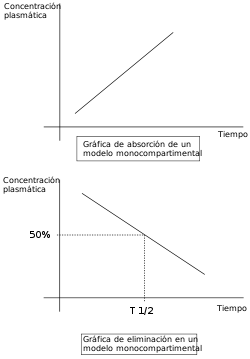 Hipótesis del transcurrir monocompartimental.
Hipótesis del transcurrir monocompartimental.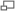
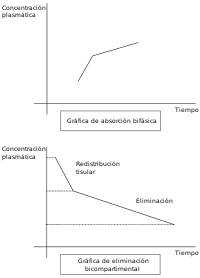
Sin embargo, no siempre estos presupuestos recogen con una fidelidad aproximada lo que ocurre realmente en el organismo. Por ejemplo, no todos los tejidos presentan la misma riqueza en aporte sanguíneo, por lo que en unos la distribución del fármaco será más lenta que en otros. Además, existen algunos tejidos (como por ejemplo el tejido del cerebro) que presentan una verdadera barrera a la llegada de los fármacos, que será saltada con mayor o menor facilidad dependiendo de las características del fármaco. De modo que, manteniendo los otros condicionantes de proporcionalidad entre los distintos tejidos y de la velocidad de eliminación, el organismo se podría comportar como dos compartimentos: uno al que podemos llamar compartimento central que presenta una velocidad de distribución más elevada y constituido por los órganos y sistemas más intensamente irrigados y un compartimento periférico constituido por los órganos menos irrigados, quedando algunos tejidos como el cerebro en una posición variable según la facilidad que presente el fármaco para atravesar la barrera que lo separa de la sangre.
Este modelo bicompartimental será diferente considerando en cual compartimento se produce la eliminación. Lo más frecuente es que la misma se produzca en el compartimento central, ya que hígado y riñones son órganos muy bien irrigados. No obstante puede darse la situación de que la eliminación se realice desde el compartimento periférico o incluso desde ambos. Obtenemos así al menos tres variedades de modelo bicompartimental, que sin embargo sigue sin explicarnos todas las posibilidades.[1]
La realidad de que algunas de las enzimas responsables del metabolismo pueden saturarse, o de la presencia de un mecanismo de eliminación activo independiente de la concentración del fármaco en el plasma, son factores que impiden la aplicación del anterior modelo. Además, la situación real es que cada tejido presenta sus propias características de distribución y que ninguna de ellas es estrictamente lineal. Si llamamos VdF al volumen de distribución del fármaco en el organismo y VdT al volumen de distribución del fármaco en un tejido dado, el primero vendrá dado por la ecuación que tenga en cuenta a todos los tejidos que actúen de forma diferente, es decir:
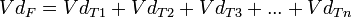
Se presenta así un modelo policomportamental, de numerosas curvas que precisaría complicadas ecuaciones para la obtención de una curva global. De hecho, existen complicados programas informáticos para dar respuesta a la misma.[1] Respuesta que de todas formas no es aún la realidad debido a la dificultad previa de encontrar los verdaderos valores de distribución del fármaco, dado que, como se verá más adelante, incluso el propio concepto de volumen de distribución es un concepto relativo que ofrece sólo un reflejo de la realidad. Por tanto, la elección del modelo va a depender de cuál sea el que ofrece el menor rango de error en función del tipo de fármaco implicado.
Modelo monocompartimental
Se le conoce como farmacocinética lineal porque al graficar la relación entre los distintos factores implicados (dosis, concentraciones en el plasma sanguíneo, eliminación, etcétera) la representación gráfica es una recta o una aproximación a ella. Es muy útil para fármacos que se distribuyen con rapidez desde el plasma a otros fluidos y tejidos.
Modelos policompartimentales
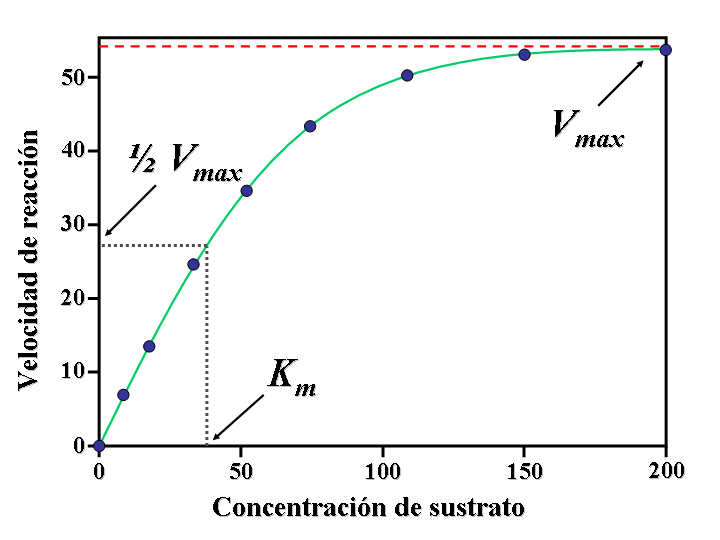
 Gráficas de absorción y eliminación bajo un modelo de farmacocinética no lineal.
Gráficas de absorción y eliminación bajo un modelo de farmacocinética no lineal.Al graficar la relación entre los distintos factores, la imagen resultante es una curva, siendo entonces necesario el cálculo de determinadas áreas bajo esa curva para hallar los resultados a las interrelaciones presentadas. Por ello, estos modelos reciben también el nombre de farmacocinética no lineal, y se basa de forma muy importante en la cinética de Michaelis-Menten. Los factores de no linealidad de una reacción, serían, entre otros, los siguientes:
- Absorción polifásica: La absorción del fármaco sigue al menos dos picos de máxima intensidad, con lo que mediatiza la linealidad de su llegada al plasma.
- La naturaleza del fármaco hace clara distinción entre tejidos de alta y baja irrigación.
- Saturación enzimática: En fármacos en los que su eliminación es dependiente de su biotransformación, al aumentar la dosis, las enzimas responsables de su metabolismo se saturan y la concentración plasmática del fármaco aumenta desproporcionalmente, por lo que su depuración deja de ser constante.
- Inducción o inhibición enzimática: Algunos fármacos tienen la capacidad de inhibir o estimular su propio metabolismo, en una reacción de retroalimentación. Tal es el caso de fluvoxamina, fluoxetina y fenitoína. Al administrar mayores dosis de estos medicamentos, las concentraciones plasmáticas de fármaco sin metabolizar aumenta y el tiempo medio de eliminación aumenta con el tiempo. Por esa razón, para fármacos con farmacocinética no-lineal, es necesario ajustar la posología o régimen en casos de incrementar la dosis.
- El riñón establece mecanismos activos de eliminación para algunos fármacos, independientes de los niveles de concentración plasmática.
Como se puede apreciar, la no linealidad puede venir determinada por razones que afectan a toda la secuencia farmacocinética: absorción, distribución, metabolismo y eliminación.
Biodisponibilidad
A efectos prácticos, se define la biodisponibilidad de un fármaco como la fracción del mismo que alcanza la circulación sistémica del paciente. O dicho de otra manera, el porcentaje de fármaco que aparece en plasma. Desde este prisma, la administración de un fármaco por vía intravenosa presentaría la mayor biodisponibilidad posible, por lo que se considera la unidad (o el 100%). A partir de aquí, la biodisponibilidad se calcula comparando la vía a estudiar con respecto a la vía intravenosa («biodisponibilidad absoluta») o a un valor estándar de otras presentaciones del fármaco en estudio («biodisponibilidad relativa»).
![B_A = \frac{[ABC]_P . D_{IV}}{[ABC]_{IV} . D_P}](/pictures/eswiki/53/5ca3c5293ac76c5cc118262203196262.png)
![\mathit B_R = \frac{[ABC]_{A} . dosis_{B}}{[ABC]_{B} . dosis_{A}}](/pictures/eswiki/56/89142147383556850b8ddeefa9d174b1.png)
Conocida la biodisponibilidad de un fármaco, podremos calcular qué modificaciones hay que realizar en su posología para alcanzar los niveles sanguíneos deseados. La biodisponibilidad es pues una razón matemática individual para cada fármaco que actúa sobre la dosis administrada. Mediante la fórmula
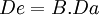 (en donde De es la dosis eficaz, B la biodisponibilidad y Da la dosis administrada) podemos calcular la cantidad de fármaco en plasma que realmente tiene capacidad para realizar su efecto.
(en donde De es la dosis eficaz, B la biodisponibilidad y Da la dosis administrada) podemos calcular la cantidad de fármaco en plasma que realmente tiene capacidad para realizar su efecto.Así, si tenemos un fármaco cuya biodisponibilidad es de 0.8 (o del 8o%) y se administra una dosis de 100 mg, la ecuación se resolvería:
De = 0,8 x 100 mg = 80 mg Es decir, de los 100 mg administrados, son realmente 80 mg los que tienen la capacidad para realizar su efecto farmacológico.
 Diferentes formas de comprimidos, los cuales conllevan diferentes comportamientos farmacocinéticos después de su administración.
Diferentes formas de comprimidos, los cuales conllevan diferentes comportamientos farmacocinéticos después de su administración.Este concepto depende de otra serie de factores inherentes a cada fármaco, como son:[2]
- Forma galénica
- Forma química
- Vía de administración
- Estabilidad
- Metabolización
Estos conceptos, que pueden verse detalladamente en el artículo principal del epígrafe, pueden cuantificarse matemáticamente y a su vez ser integrados para obtener una ecuación matemática de los mismos:
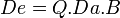
,
donde Q sería la constante de pureza del fármaco.[2]
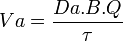
siendo Va la velocidad de administración del fármaco y τ la constante que representa la velocidad a la que el fármaco absorbido alcanza la circulación sistémica.
Finalmente, por la ecuación de Henderson-Hasselbalch, y sabiendo el
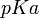 del fármaco (pH al cual presenta equilibrio entre sus moléculas ionizadas y no ionizadas), podemos calcular la cantidad de fármaco no ionizado, y, por tanto, la cantidad de fármaco objeto de la absorción:
del fármaco (pH al cual presenta equilibrio entre sus moléculas ionizadas y no ionizadas), podemos calcular la cantidad de fármaco no ionizado, y, por tanto, la cantidad de fármaco objeto de la absorción: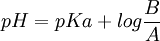
Cuando dos fármacos tienen la misma biodisponibilidad se dice que son equivalentes biológicos o bioequivalentes. Este concepto de bioequivalencia es importante porque en la actualidad es la vara de medir para la autorización de los medicamentos genéricos en numerosos países.
El acrónimo LADME
Una vez que el fármaco entra en contacto con el organismo, suceden varias fases que se reconocen con el acrónimo LADME:
- Liberación de la sustancia activa,
- Absorción de la misma por parte del organismo
- Distribución por el plasma y los diferentes tejidos,
- Metabolización, es decir inactivación de una sustancia xenobiótica y, finalmente,
- Excreción o eliminación de la sustancia o de los productos de su metabolismo.
No obstante, muchos manuales engloban la primera fase dentro de la segunda, ya que en numerosas ocasiones se administra el fármaco en forma de principio activo, con lo que ésta fase no existe. Otros hablan de una fase que engloba la distribución, metabolización y excreción que sería la «fase de disposición». Finalmente también hay autores que incluyen el aspecto toxicológico de cada fármaco en lo que se conocería como ADME-Tox o ADMET.
Cada una de las fases está sujeta a las interacciones físico-químicas entre fármaco y organismo, que se pueden expresar de forma matemática. La farmacocinética, pues, se apoya en ecuaciones matemáticas que permiten predecir el comportamiento del fármaco, y que dan cuenta, de una forma preferente, de la relación que existe entre las concentraciones plasmáticas y el tiempo transcurrido desde la administración.
Liberación
La liberación es el primer paso del proceso en el que el medicamento entra en el cuerpo y libera el contenido del principio activo administrado. El fármaco debe separarse del vehículo o del excipiente con el que ha sido fabricado, y para algunos autores comprende tres pasos: desintegración, disgregación y disolución. Se hace una especial referencia a la ionización de las moléculas del fármaco como factor limitante de la absorción, debido a las propiedades de las membranas celulares que dificultan el paso a su través de moléculas ionizadas. La recomendación de masticar los comprimidos o tabletas realizada por muchos profesionales radica, precisamente, en facilitar esta fase, en concreto la disgregación.
En todo caso, es necesario recordar que las características de los excipientes tienen un papel fundamental, ya que tienen como una de sus funciones el crear el ambiente adecuado para que el fármaco se absorba correctamente. Es por ello que medicamentos con la misma dosis, pero de distintas marcas comerciales pueden tener distinta bioequivalencia, es decir, alcanzan concentraciones plasmáticas distintas, y, por tanto, efectos terapéuticos diferentes.
Disolución
En una situación típica, al ingerir una tableta pasa por el esófago al estómago. Por razón de que el estómago tiene un ambiente acuoso, es el primer lugar donde la tableta se disolverá. La velocidad de disolución es un elemento clave en el control de la duración del efecto del fármaco, y por ello, diferentes formas del mismo medicamento pueden tener los mismos ingredientes activos, pero difieren en la velocidad de disolución. Si se administra un fármaco bajo una forma galénica que no es rápidamente disuelta, el fármaco se absorberá más gradualmente en el tiempo, alcanzando una más larga duración en su acción. La consecuencia es una mejora en su complianza, logrando en definitiva, que el medicamento no tenga que ser tomado tan a menudo. Además, una forma de liberación lenta mantendrá concentraciones en rangos terapéuticos aceptables por un período de tiempo más duradero a diferencia de las presentaciones de liberación rápida, que tienen picos de concentraciones plasmáticas más pronunciados.
La velocidad de disolución se describe por la ecuación de Noyes-Whitney:
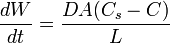
Donde:
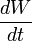 es la velocidad de disolución.
es la velocidad de disolución.- A es la área superficial del sólido.
- C es la concentración del sólido en el medio de disolución principal.
- Cs es la concentración del sólido en la capa de difusión que rodea al sólido.
- D es el coeficiente de difusión.
- L es el grosor de la capa de difusión.
Debido a que las soluciones ya están en un estado disuelto, no tienen necesidad de pasar por una etapa de disolución antes de que se comience su absorción.
Ionización
Las membranas celulares presentan una resistencia al paso de moléculas ionizadas superior a la que presenta a las sustancias no ionizadas y liposolubles. Este hecho es de importancia sobre todo con sustancias que son anfotéricamente débiles. El pH ácido del estómago y la posterior alcalinización del mismo en el intestino, modifican los grados de ionización de ácidos y bases débiles, dependiendo del pKa de cada sustancia.[3] El pKa es el pH en el que una sustancia presenta un equilibrio entre las moléculas ionizadas y las no ionizadas, y para su cálculo es importante considerar la ecuación de Henderson-Hasselbalch.
Absorción
La absorción significa atravesar algún tipo de barrera, diferente según la vía de administración usada, pero que en último término se puede reducir al paso de barreras celulares. O dicho de otra forma, la interacción de la molécula con una membrana biológica, donde las características fisicoquímicas, tanto del fármaco como de la membrana, determinarán el resultado del proceso.
Membranas biológicas
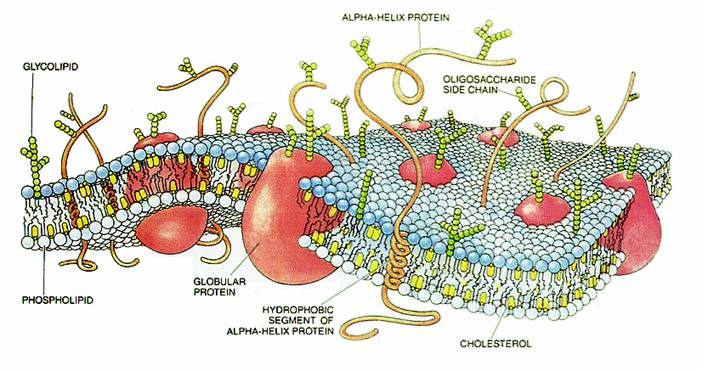
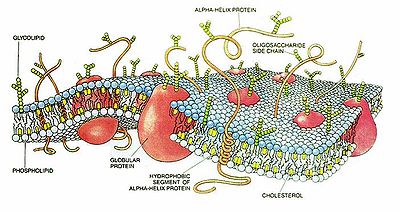 Esquema de una membrana celular
Esquema de una membrana celularEs indispensable conocer la estructura de la membrana citoplasmática debido a su estrecha e importante relación con la farmacocinética, que implica el pasaje de las drogas a través de las membranas. La membrana citoplasmática consiste en una capa bimolecular de lípidos, con moléculas de proteínas intercaladas, que adquiere un espesor de 75 a 80 Å (angstrom, unidad de longitud).[4]
Los fosfolípidos son responsables de las características de permeabilidad de la membrana así como eslabón importante en la cadena anabólica de numerosas sustancias de defensa (prostaglandinas, leucotrienos, ...). Suponen aproximadamente un 40% a 45% de los componentes de la membrana.
Por su parte, las proteínas constituyen alrededor del 50% de los constituyentes de las membranas, y le dan la rigidez estructural necesaria a la misma. Además, se comportan como el punto de inicio de las reacciones a las moléculas que llegan hasta la membrana (receptores), las metabolizan (enzimas), transportan moléculas en contra del gradiente de concentración a ambos lados de la membrana (bombas), o crean canales por donde puedan pasar éstas moléculas (proteínas canal).
Finalmente, nos podemos encontrar entre un 7% y un 10% de hidratos de carbono (glucolípidos y glucoproteínas) que actúan como modulador de las proteínas receptores.
El receptor celular es el punto último del viaje del fármaco destinado a lograr un efecto sobre el organismo humano. De las complejas interrelaciones entre ambos se encarga otra disciplina de la farmacología: la farmacodinámica.
Vías de administración
Las barreras que ha de atravesar y las características de la absorción de cada sustancia vienen determinadas por cual haya sido la vía por la que ha llegado la misma a entrar en contacto con el organismo, o dicho de otro modo, de cual sea la vía de administración. Aquí se verá sólo una breve tabla de las diferentes vías de administración, con las características especiales en cada caso de cara a la absorción.
La vía oral es la vía recomendada para humanos. Desafortunadamente, no todos los productos pueden adaptarse para su uso por esta vía. En la vía oral el fármaco llega al organismo habitualmente después de la deglución. Una vez en el estómago, se somete a las características de los jugos del mismo, que por su acidez favorece mucho la ionización del fármaco, lo que hace que la absorción sea difícil. A pesar de todo, no son escasos los fármacos que se absorben a nivel de la mucosa gástrica: los muy liposolubles, como el alcohol o ácidos débiles como los salicilatos o los barbitúricos que presentan menores niveles de ionización a pH bajo. Cuando llega el fármaco al intestino delgado cambia el pH luminal y se favorece bastante la absorción pasiva. De hecho, prácticamente todos los fármacos, menos los ácidos y bases fuertes, se absorben a este nivel. Además, en la mucosa intestinal hay numerosos mecanismos para realizar procesos de absorción en contra de gradiente, aunque difícilmente se logran niveles plasmáticos suficientes para que sean efectivos. Esta falta de absorción para algunos fármacos se aprovecha para utilizarlos a nivel local (como la neomicina o los laxantes). Igualmente, por su similitud estructural, se utiliza este efecto para administrar fármacos que no atraviesen la piel y que actúen a nivel local, constituyendo lo que se conoce como vía dérmica o vía tópica.
La vía parenteral ofrece indudables ventajas sobre la vía oral: permite su uso en pacientes que no pueden o no deben deglutir, permite el uso de sustancias polipeptídicas y otras que se inactivan por los jugos gastrointestinales y evitan el primer paso hepático. Sin embargo precisa de instrumental para su realización y presenta inconvenientes como la infección local, tromboflebitis, neuralgias, necrosis dérmicas, etc. Desde el punto de vista farmacodinámico, la principal ventaja es la facilidad para ajustar la dosis eficaz, ya que la biodisponibilidad se considera del 100% en la mayoría de los casos.
Respecto a la vía respiratoria su interés fundamental es que brinda la posibilidad de la utilización de sustancias en estado gaseoso (casi exclusivamente oxígeno o anestésicos generales). La absorción sigue las leyes del intercambio de gases a nivel alveolar y tiene la ventaja de poner en disposición una gran superficie de absorción.[3]
Características de la absorción
Hay que tener presente la existencia de una serie de factores que modifican la absorción:
- Solubilidad: la absorción del fármaco en más rápida cuando está en solución acuosa con respecto a si está en solución oleosa, y, a su vez, ambas son más rápidas que la que presentaría en forma sólida.
- Cinética de disolución de la forma farmacéutica del medicamento. De la misma depende la velocidad y la magnitud de la absorción del principio activo.
- Concentración del fármaco: a mayor concentración, mayor absorción.
- Circulación en el sitio de absorción: a mayor circulación, mayor absorción.
- Superficie de absorción: a mayor superficie, mayor absorción.
Teniendo en cuenta estos factores, los mecanismos por los cuales, independientemente de la vía usada, se produce la absorción son los siguientes:
Absorción pasiva o difusión pasiva
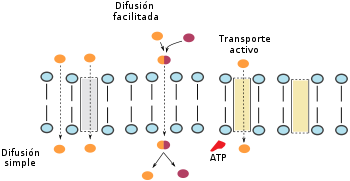 Mecanismos de absorción.
Mecanismos de absorción.El paso de la sustancia implicada se produce sin gasto de energía, a favor de gradientes de concentración. Puede producirse a través de la membrana propiamente dicha o a través de ciertas proteínas que forman poros.
- Difusión simple: depende del tamaño de las moléculas, y puede realizarse a través de la bicapa lipídica de la membrana o a través de los poros acuosos constituidos por las proteínas insertas en la misma. Las sustancias no ionizadas tienen mayor facilidad para la misma, siguiendo la ley de Fick, por la cual
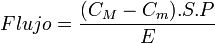
En donde C representa las concentraciones a ambos lados de la membrana, S es el área de interacción, P el coeficiente de permeabilidad y E el espesor de la membrana. Ecuación de donde se deduce que las sustancias tienden a ionizarse cuando el medio muestra un pH contrario a su naturaleza.
- Difusión facilitada: se debe a la presencia de un gradiente a ambos lados de la membrana para otras moléculas que tienen la propiedad de unirse al fármaco y arrastrarlo en su migración. Son las moléculas facilitadoras, y se incluyen dentro de la difusión pasiva debido a que no consumen energía en su trasiego. Sin embargo, a diferencia de la difusión simple, este mecanismo es saturable, al depender del número de moléculas facilitadoras.
Absorción activa o transporte activo
El paso de la sustancia implica un gasto energético en forma de moléculas de ATP. Permite la absorción contra gradiente y depende también de las moléculas facilitadoras, que en esta ocasión no migran en función de un gradiente, sino gracias al gasto energético. Por tanto es un mecanismo también saturable. Se realiza mediante las proteínas bomba de la membrana (ATP Binding Cassete), teniendo especial transcendencia la MDR1(del inglés MultiDrug Resistence tipo 1) que exporta un gran número de fármacos y es factor clave de la resistencia de las células cancerosas a los quimioterápicos.[5]
La endocitosis es un mecanismo propio de algunas células por el que mediante la formación de vesículas originadas a partir de la membrana citoplásmica, introducen en su interior sustancias externas a ellas. Es un mecanismo que consume gran cantidad de energía, pero tiene la ventaja de introducir grandes cantidades de material al interior celular.
Distribución
La distribución de los fármacos puede definirse, entre otras formas, como la llegada y disposición de un fármaco en los diferentes tejidos del organismo. Es un proceso muy importante, toda vez que, según su naturaleza, cada tejido puede recibir cantidades diferentes del fármaco, el cual, además, pasará allí tiempos variables.[6]
A la hora de hablar de la distribución, habrá que tener en cuenta los conceptos sobre compartimentación del organismo vistos en el apartado de Modelos farmacocinéticos.
Factores que afectan la distribución
Son múltiples, pero siguiendo a Pascuzzo, los más importantes son los tres siguientes: los volúmenes físicos del organismo, la tasa de extracción y la unión a proteínas plasmáticas y, o, tisulares.
Volúmenes físicos del organismo
Este concepto está relacionado con la multicompartimentalización. Considerando los fármacos como solutos, los distintos tejidos con especificidad del organismo van a actuar como los solventes que darán pie a las diferentes concentraciones del fármaco. Así, dependiendo de la naturaleza química de éste, habrá una especial predisposición de las sustancias liposolubles por la grasa corporal o de las hidrosolubles por el líquido extracelular. Este volumen de distribución (Vd) de un fármaco en el organismo es tan sólo aparente, pues conceptualmente se trataría del volumen necesario para contener de forma homogénea en todo el organismo una cantidad determinada de fármaco, que viene dada por el nivel de la concentración del mismo en el plasma. Desde el punto de vista físico el Vd viene determinado por la siguiente fórmula:
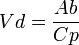 en donde Ab es la cantidad total de fármaco en el cuerpo y Cp la concentración plasmática del mismo.
en donde Ab es la cantidad total de fármaco en el cuerpo y Cp la concentración plasmática del mismo.Siendo la Ab conocida, pues es equivalente a la dosis de fármaco administrada, la fórmula nos indica que la relación existente entre Vd y la Cp es una relación de proporcionalidad inversa. Es decir, que a mayor Cp menor Vd y viceversa. O lo que es lo mismo, que los factores que aumenten la Cp disminuirán el valor del Vd. Esto nos pone sobre la pista de la importancia del conocimiento de las concentraciones plasmáticas del fármaco y de los factores que lo modifican.
Aplicando a esta fórmula los conceptos aprendidos en el apartado de la biodisponibilidad, podemos calcular la cantidad de fármaco a administrar para conseguir una determinada concentración de fármaco en el organismo (dosis de carga):
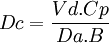
Este concepto tiene interés clínico, pues a veces necesitamos alcanzar una determinada concentración de fármaco que sabemos es la óptima para que realice sus efectos en el organismo (caso de la digitalización de un paciente).
Tasa de extracción
Se refiere a la proporción del fármaco que es retirado de la circulación por cada órgano, una vez que el flujo sanguíneo lo haya hecho pasar a través de dicho órgano.[6] Este nuevo concepto integra otros anteriores, ya que la tasa de extracción va a depender de distintos factores:
- Características del fármaco, entre ellas su pKa.
- Redistribución tisular: En algunos fármacos se produce una distribución rápida e intensa en determinados tejidos, hasta llegar al equilibrio con la concentración plasmática. Sin embargo otros tejidos más lentos continúan retirando fármaco del plasma, con lo que la concentración en el primer tejido queda por encima de la plasmática y por tanto sale fármaco del tejido hacia el plasma. Este fenómeno se sigue sucediendo durante un tiempo hasta alcanzar el equilibrio definitivo. Se obtiene por tanto dos concentraciones del fármaco en el tejido más sensible: una inicial más elevada y otra posterior consecuencia de la redistribución tisular.
- Diferencial de concentración con los tejidos.
- Superficie de intercambio.
- Presencia de barreras naturales. Son obstáculos a la difusión similares a las encontradas en la absorción. Las más interesantes son:
- Permeabilidad de los lechos capilares, que no es igual en todos los tejidos.
- Barrera hematoencefálica: está localizada entre el plasma sanguíneo de los vasos cerebrales y el espacio extracelular del encéfalo. Dificulta la llegada de fármacos al mismo.
- Barrera placentaria: en la mujer embarazada, evita la llegada de gran cantidad de fármacos al feto, que pudieran ser tóxicos para el mismo.
Unión a proteínas plasmáticas
Algunos fármacos tienen la capacidad de unirse a distintos tipos de proteínas vehiculizadas en el plasma sanguíneo. Esto es de gran importancia dado que, como sabemos, sólo el fármaco que se encuentra diluido en el plasma será capaz de pasar a los tejidos. De esta manera la unión del fármaco a las proteínas plasmáticas actúa como un reservorio del mismo dentro del organismo y disminuye las concentraciones finales en los tejidos. La unión de fármacos y proteínas es poco específica y usualmente lábil y reversible, generalmente a través de enlaces iónicos, puentes de hidrógeno, fuerzas de Van der Waals y, con menos frecuencia, enlaces covalentes. Esto implica que un fármaco puede ser desplazado de su unión a la proteína por otra sustancia (u otro fármaco) y que en todo caso, la unión está sujeta a saturación. También, existe un equilibrio entre el fármaco libre en el plasma y el unido a proteínas, por lo que la proporción de fármaco unido a las mismas es estable, independientemente de su cantidad total en el plasma.
Por estudios realizados in vitro en condiciones ideales, el equilibrio entre la concentración plasmática y tisular del fármaco sólo se ve alterado de forma significativa con índices de fijación a proteínas plasmáticas superiores al 90%. A partir de estos niveles se produce un "secuestro" del fármaco que disminuye su presencia en los tejidos por debajo del 50% del total. Esto es importante a la hora de considerar las interacciones farmacológicas: un fármaco con un índice de fijación a proteínas plasmáticas inferior al 90%, si es desplazado de su unión a las proteínas por otro fármaco no va a aumentar significativamente su presencia en los tejidos. Por el contrario, con índices de unión a proteínas plasmáticas superiores al 95%, pequeños desplazamientos pueden originar importantes modificaciones de la concentración tisular y, por tanto, mayor riesgo de toxicidad por exceso de su efecto en los tejidos.
De las proteínas plasmáticas quizás las de más interés sean las albúminas, por su cantidad y su capacidad para unirse a otras sustancias. Otras proteínas de interés son las glicoproteínas, las lipoproteínas y en menor medida las globulinas.
Como podrá comprenderse, situaciones clínicas que supongan modificación de los niveles de proteínas plasmáticas (por ejemplo hipoalbuminemias secundarias a procesos renales) pueden tener transcendencia en el efecto y toxicidad de un fármaco que presente índices de unión a proteínas plasmáticas superiores al 90% (ó 0,9).
Metabolismo o biotransformación
Muchos fármacos son transformados en el organismo debido a la acción de enzimas. Esta transformación, destinada a contrarrestar el posible efecto perjudicial de una sustancia extraña al organismo, es el concepto básico del metabolismo xenobiótico, siendo los fármacos las sustancias xenobióticas por excelencia.
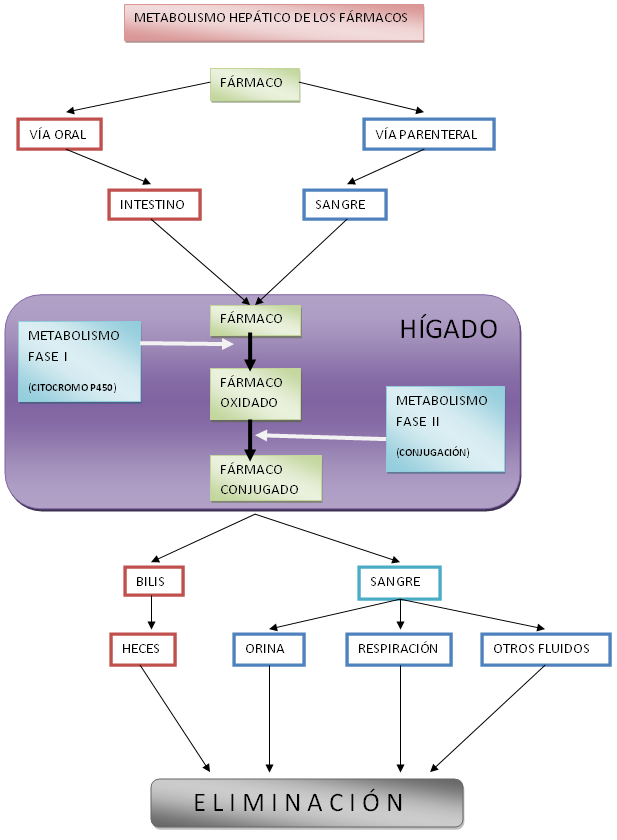
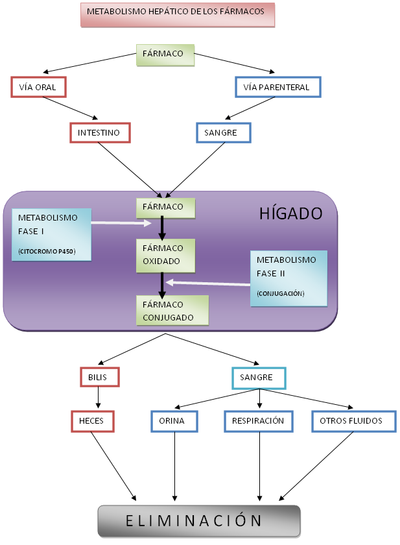 Diagrama del metabolismo hepático de fase I y II
Diagrama del metabolismo hepático de fase I y IILa transformación puede consistir en la degradación (oxidación, reducción o hidrólisis), donde el fármaco pierde parte de su estructura, o en la síntesis de nuevas sustancias con el fármaco como parte de la nueva molécula (conjugación). La oxidación se realiza fundamentalmente por las isoenzimas del citocromo P450 en lo que se conoce como metabolismo de fase I. La conjugación es la fase fundamental del metabolismo de fase II, pudiendo existir una tercera fase o metabolismo de fase III, característica de los organismos pluricelulares.
En el humano y en la mayoría de los mamíferos, el metabolismo de los fármacos se realiza fundamentalmente a nivel del hígado Como resultado de la biotransformación se obtienen nuevas sustancias que reciben el nombre de metabolitos. Los metabolitos pueden mantener la capacidad del fármaco original para ejercer sus efectos, o bien haberla vista disminuida, aumentada o incluso haber cambiado sus efectos por otros distintos. Por ello se habla de metabolitos activos, o inactivos. Incluso, en ocasiones el fármaco no presenta actividad farmacológica alguna, siendo alguno de sus metabolitos los que realmente ejercen su actividad. Se habla en este caso de profármacos, y un ejemplo claro son algunas estatinas (simvastatina y lovastatina). Evidentemente, los profármacos dependen del buen funcionamiento del metabolismo para poder ejercer de forma adecuada sus efectos.
En ocasiones los propios fármacos o algunos de sus metabolitos son capaces de modificar la capacidad metabólica de las enzimas, aumentando o disminuyendo su actividad. Esta inducción o inhibición enzimática conlleva una mejoría o empeoramiento de la depuración de los fármacos, y subsecuentemente un posible aumento de su toxicidad o bien una disminución de su efecto. Este fenómeno es de gran trascendencia para algunas isoenzimas del citocromo p450, siendo objeto de continua investigación la determinación de los sustratos y de los inductores o inhibidores de las mismas.
La dotación enzimática viene determinada de forma genética, existiendo diferentes niveles de actividad en función del genotipo. Un ejemplo son los acetiladores lentos: sujetos que poseen una carga enzimática con menor capacidad para la metilación, por lo que en ellos son más frecuentes las interacciones y los casos de reacción adversa al fármaco. Estos son casi el 90% de la población japonesa, mientras que entre los europeos o los africanos están equilibrados con los acetiladores rápidos. Otros ejemplos pueden ser los metiladores rápidos, intermedios o lentos.
La farmacocinética estudia los mecanismos mediante los cuales se producen estas transformaciones, los tejidos en que ocurre, la velocidad de estos procesos y los efectos de las propias drogas y sus metabolitos sobre los mismos procesos enzimáticos. A modo de ejemplo, véase el diagrama del metabolismo hepático de los fármacos.
Excreción
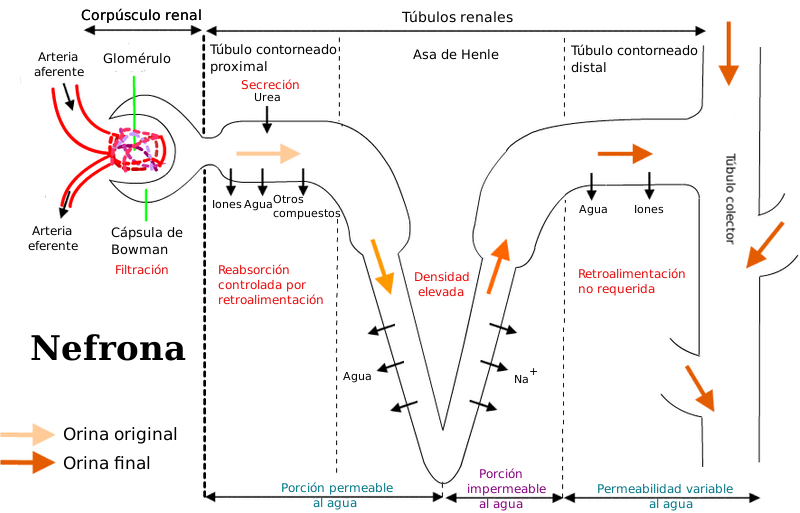
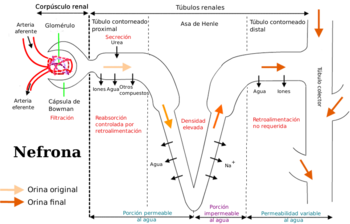 Identificación del flujo renal a lo largo de la nefrona.
Identificación del flujo renal a lo largo de la nefrona.Los fármacos son eliminados del organismo inalterados (moléculas de la fracción libre) o modificados como metabolitos a través de distintas vías. El riñón es el principal órgano excretor, aunque existen otros, como el hígado, la piel, los pulmones o estructuras glandulares, como las glándulas salivales y lagrimales. Estos órganos o estructuras utilizan vías determinadas para expulsar el fármaco del cuerpo, que reciben el nombre de vías de eliminación:
- Orina,
- Lágrimas,
- Sudor
- Saliva
- Respiración
- Leche materna
- Heces
- Bilis
En lo que respecta al riñón, los fármacos son excretados por filtración glomerular y por secreción tubular activa siguiendo los mismos pasos y mecanismos de los productos del metabolismo intermedio. Así, las drogas que filtran por el glomérulo sufren también los procesos de la reabsorción tubular pasiva. Por filtración glomerular solo se eliminan las drogas o los metabolitos no ligados a las proteicas plasmáticas (fracción libre), y muchos otros (como los ácidos orgánicos) son secretados activamente. En los túbulos proximal y distal las formas no ionizadas de ácidos o bases débiles son reabsorbidas pasiva y activamente. Cuando el fluido tubular se hace más alcalino, los ácidos débiles se excretan más fácilmente y esto disminuye la reabsorción pasiva. Lo inverso ocurre con las bases débiles. Por eso en algunas intoxicaciones puede incrementarse la eliminación del fármaco tóxico, alcalinizando la orina y forzando la diuresis.
En otras ocasiones los fármacos son eliminados en la bilis con la que llegan hasta el intestino. Allí se unen a la fracción no absorbida del fármaco y se eliminan con las heces o bien pueden sufrir un nuevo proceso de absorción y ser eliminados finalmente por el riñón.
Las otras vías tienen poca transcendencia, salvo para fármacos muy concretos, como la vía respiratoria para el alcohol o los gases anestésicos, aunque en el caso de la leche materna es de especial trascendencia. El recién nacido presenta todavía cierta inmadurez de hígado o riñones y es más sensible a los efectos tóxicos del fármaco. Por ello hay que conocer qué fármacos pueden eliminarse a través de la leche materna para evitarlos.
Parámetros farmacocinéticos de la excreción
La farmacocinética estudia la forma y velocidad de depuración de los fármacos y sus metabolitos por los distintos órganos excretores, en relación con las concentraciones plasmáticas del fármaco. Para ello precisa de la definición operativa de algunos conceptos relativos a la excreción.
Vida media
La vida media plasmática o vida media de eliminación es el tiempo necesario para eliminar el 50% del fármaco del organismo. O bien, el tiempo que tarda la concentración plasmática del fármaco en reducirse a la mitad de sus niveles máximos.
Aclaramiento
Al medir la concentración plasmática de un fármaco antes de pasar por un órgano (sangre arterial) y después de haber pasado por él (sangre venosa) si se encuentra una diferencia de concentraciones se puede deducir que el órgano ha eliminado una parte del fármaco, aclarando la concentración del mismo. Desde esta óptica, se considera el aclaramiento como el volumen plasmático libre totalmente de fármaco por unidad de tiempo, por lo que se mide en unidades de volumen por unidades de tiempo. El aclaramiento, puede determinarse de una forma global («aclaramiento sistémico») o de forma individualizada para cada vía (aclaramiento hepático, renal, etc.). La ecuación que recoge este concepto sería:
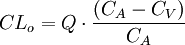
En donde CLo es el aclaramiento del órgano, CA la concentración plasmática en sangre arterial, CA la concentración plasmática en sangre venosa y Q el flujo sanguíneo del órgano.
Es fácil comprender que cada órgano tendrá sus condicionantes del aclaramiento, en función de su mecanismo de acción para realizar la depuración. En lo que respecta al «aclaramiento renal», viene determinado por factores como el grado de unión a proteínas plasmáticas del fármaco (sólo se filtra el fármaco libre), saturación de los transportadores (la secreción activa depende de proteínas transportadoras, que son saturables), o el número de nefronas funcionantes (de donde la importancia de situaciones como la insuficiencia renal).
En el caso del hígado, el «aclaramiento hepático» es fruto del metabolismo y por tanto está determinado por los factores que alteran el mismo así como por la cantidad de hepatocitos funcionantes, lo que justifica la importancia clínica de la insuficiencia hepática.
Estado de equilibrio
El estado de equilibrio o concentración estable es aquel en el que los aportes plasmáticos de fármaco se equilibran con la eliminación del mismo. Es fundamental su cálculo para decidir el período entre dosis y la cantidad de fármaco en cada una de ellas, en tratamientos prolongados.
Otros parámetros
Otros parámetros de interés y ya vistos son la biodisponibilidad o el volumen aparente de distribución.
Farmacocinética Clínica
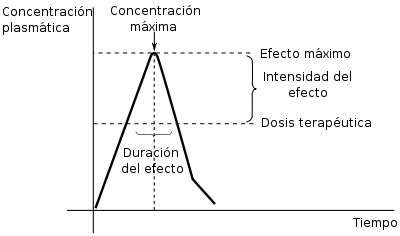 Gráfica básica para valorar las implicaciones terapéuticas de la farmacocinética.
Gráfica básica para valorar las implicaciones terapéuticas de la farmacocinética.La Farmacocinética Clínica resulta de la aplicación directa en los pacientes de los conocimientos farmacocinéticos del fármaco en cuestión y de las características de la población a la que pertenece (o puede adscribirse) el paciente en concreto.
Gracias a la Farmacocinética Clínica, por ejemplo, se relanzó el empleo de ciclosporina como tratamiento inmunosupresor para posibilitar el trasplante de órganos sólidos (como el riñón), dado que tras demostrarse inicialmente sus propiedades terapéuticas se descartó prácticamente su uso por la nefrotoxicidad que provocaba en numerosos pacientes.[7] Una vez que se comprobó que se podía individualizar la posología de la ciclosporina analizando las concentraciones plasmáticas de cada paciente (monitorización farmacocinética), la seguridad de este fármaco ha posibilitado gran cantidad de abordajes de trasplantes.
Clínicamente, la monitorización suele realizarse mediante la determinación de las concentraciones plasmáticas, ya que suele ser la determinación más accesible y fiable de las disponibles. Los principales criterios para determinar las concentraciones plasmáticas de un fármaco son:[8]
- Estrecho intervalo terapéutico (intervalo entre las concentraciones tóxica y terapéutica)
- Alta toxicidad
- Elevado riesgo vital.
Algunos fármacos en los que se recomienda la monitorización farmacocinética son:
Fármacos con indicación de monitorización. - + Fenitoína
- + Carbamazepina
- + Ácido Valproico
- + Lamotrigina
- + Etosuximida
- + Fenobarbital
- + Primidona
- Medicamentos cardioactivos
- Medicamentos inmunosupresores
- + Ciclosporina
- + Tacrolimus
- + Sirolimus
- + Everolimus
- + Micofenolato
- Medicamentos antibióticos
- + Gentamicina
- + Tobramicina
- + Amikacina
- + Vancomicina
- Medicamentos broncodilatadores
- Medicamentos citostáticos
- + Metotrexato
- + 5-Fluoruracilo
- + Irinotecan
* Medicamentos antivirales (VIH) - + Factor VIII,
- + Factor IX,
- + Factor VIIa,
- + Factor XI
Véase también
- Cinética enzimática
- Farmacodinámica
- Farmacometría
- Idiosincrasia farmacológica
- Interacción farmacológica
- Farmacia
- Bioequivalencia
- Medicamentos Similares
Referencias
- Alberts et al, Introducción a la Biología Celular, pág. 375-376, 2ª edición, Ed. Médica Panamericana
- Alberts et al, Biología Molecular de la célula, pág. 595, 4ª edición, Ed. Omega
- Armijo JA. 2003. Farmacocinética: Absorción, Distribución y Eliminación de los Fármacos. En: Flórez J, Armijo JA, Mediavilla A, Farmacología Humana, 4ta edición. Masson. Barcelona. pp: 51-79.
- Balani SK, Miwa GT, Gan LS, Wu JT, Lee FW., Strategy of utilizing in vitro and in vivo ADME tools for lead optimization and drug candidate selection, Curr Top Med Chem. 2005;5(11):1033-8.
- Beal, S.; Sheiner L.B. "The NONMEM System". The American Statistician 34: 118–9. (1980). doi:10.2307/2684123.
- Cooper, La célula, pág 470-471, 2ª edición, Ed. Marbán
- Covey TR, Lee ED, Henion JD "High-speed liquid chromatography/tandem mass spectrometry for the determination of drugs in biological samples". Anal. Chem. 58: 2453–60. (October 1986). doi:10.1021/ac00125a022. PMID 3789400.
- Covey TR, JB Crowther, EA Dewey, JD Henion "Thermospray liquid chromatography/mass spectrometry determination of drugs and their metabolites in biological fluids". Anal. Chem. 57 (2): 474–81. (February 1985). doi:10.1021/ac50001a036. PMID 3977076.
- Danielson P (2002). "The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humans". Curr Drug Metab 3 (6): 561-97. PMID 12369887.
- Davies K (1995). "Oxidative stress: the paradox of aerobic life". Biochem Soc Symp 61: 1-31. PMID 8660387.
- Devlin, T. M. 2004. Bioquímica, 4ª edición. Reverté, Barcelona. ISBN 84-291-7208-4
- Galvão T, Mohn W, de Lorenzo V (2005). "Exploring the microbial biodegradation and biotransformation gene pool". Trends Biotechnol 23 (10): 497-506. PMID 16125262.
- Hsieh Y, Korfmacher WA "Increasing speed and throughput when using HPLC-MS/MS systems for drug metabolism and pharmacokinetic screening". Current Drug Metabolism 7 (5): 479–89.(June 2006). PMID 16787157. Disponible en [4]
- Janssen D, Dinkla I, Poelarends G, Terpstra P (2005). "Bacterial degradation of xenobiotic compounds: evolution and distribution of novel enzyme activities". Environ Microbiol 7 (12): 1868-82. PMID 16309386.
- Kathleen Knights; Bronwen Bryant (2002). Pharmacology for Health Professionals. Amsterdam: Elsevier. ISBN 0-7295-3664-5.
- King C, Rios G, Green M, Tephly T (2000). "UDP-glucuronosyltransferases". Curr Drug Metab 1 (2): 143-61. PMID 11465080.
- Malcolm Rowland, Thomas N. Tozer. "Farmacocinética clínica: Conceptos y Aplicaciones"
- Pharmacokinetics. (2006). En Mosby's Dictionary of Medicine, Nursing, & Health Professions. Philadelphia, PA: Elsevier Health Sciences. Disponible en [5] Última visita 11, diciembre de 2008,
- Sheehan D, Meade G, Foley V, Dowd C (2001). "Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily". Biochem J 360 (Pt 1): 1-16. PMID 11695986.
- Sheiner, L.B.; Beal, S.L., Rosenberg, B. Marathe, V.V. "Forecasting Individual Pharmacokinetics". Clin. Pharmacol. Ther. 26: 294–305. (1979). PMID 466923.
- Sheiner, L.B.; Rosenberg, B., Marathe, V.V. "Estimation of Population Characteristics of Pharmacokinetic Parameters from Routine Clinical Data". J. Pharmacokin. Biopharm. 5: 445–79. 1.997 doi:10.1007/BF01061728.
- Sies H (1997). "Oxidative stress: oxidants and antioxidants". Exp Physiol 82 (2): 291-5. PMID 9129943.
- Singh SS., Preclinical pharmacokinetics: an approach towards safer and efficacious drugs, Curr Drug Metab. 2006 Feb;7(2):165-82.
- Testa B, Krämer S (2006). "The biochemistry of drug metabolism--an introduction: part 1. Principles and overview". Chem Biodivers 3 (10): 1053-101. PMID 17193224.
- Tetko IV, Bruneau P, Mewes HW, Rohrer DC, Poda GI., Can we estimate the accuracy of ADME-Tox predictions?, Drug Discov Today. 2006 Aug;11(15-16):700-7, pre-print.
- Tu B, Weissman J (2004). "Oxidative protein folding in eukaryotes: mechanisms and consequences". J Cell Biol 164 (3): 341-6. PMID 14757749.
- Vertuani S, Angusti A, Manfredini S (2004). "The antioxidants and pro-antioxidants network: an overview". Curr Pharm Des 10 (14): 1677-94. PMID 15134565.
- ↑ a b Milo Gibaldi, Donald Perrier. FarmacocinéticaReverté 1982 pg 1-10. ISBN 84-291-5535-X, 9788429155358
- ↑ a b Michael E. Winter, Mary Anne Koda-Kimple, Lloyd Y. Young, Emilio Pol Yanguas Farmacocinética clínica básica Ediciones Díaz de Santos, 1994 pg 8-14 ISBN 84-7978-147-5, 9788479781477
- ↑ a b Simonetta Baroncini, Antonio Villani, Gianpaolo Serafini Anestesia neonatal y pediátrica (en español). Publicado por Elsevier España, 2006; pág 19. ISBN 84-458-1569-5
- ↑ Malgor - Valsecia Farmacología general: Farmacocinética. Disponible en [1] Última visita el 10 de enero del 2009.
- ↑ Raisman JS, Gonzalez, AM. Célula eucariota. Transporte activo y pasivo. Disponible en [2]. Última visita 10 enero de 2009
- ↑ a b Carmine Pascuzzo Lima. Farmacocinética III:Distribución Disponible en [3] Visitado el 10 de enero de 2009
- ↑ R. García del Moral, M. Andújar y F. O'Valle Mecanismos de nefrotoxicidad por ciclosporina A a nivel celular (artículo completo disponible en español). NEFROLOGIA. Vol. XV. Suplemento 1, 1995. Consultado 23 de febrero de 2008.
- ↑ Joaquín Herrera Carranza Manual de farmacia clínica y Atención Farmacéutica (en español). Publicado por Elsevier España, 2003; pág 159. ISBN 84-8174-658-4
Enlaces externos
- http://www.pharmer.com Pharmaceutical
- http://www.pkwiki.com
- http://vam.anest.ufl.edu/demos/onecompbolus.html
- http://www.thermo.com/pkpd
- http://www.biokinetica.pl
- http://www.biokinetica.pl/farmakokinetyka.pdf
- http://cti.itc.virginia.edu/~cmg/Demo/scriptFrame.html
Categorías: Bioquímica clínica | Farmacología | Farmacia



Wikimedia foundation. 2010.