- Síndrome de Bardet-Biedl
-
Síndrome de Bardet-Biedl 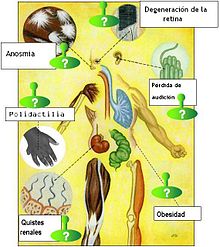
Síntomas más comunes de la enfermedad de Bardet-BiedlClasificación y recursos externos CIE-10 Q87.8 CIE-9 759.89 OMIM 209900 DiseasesDB 7286 MeSH D020788  Aviso médico
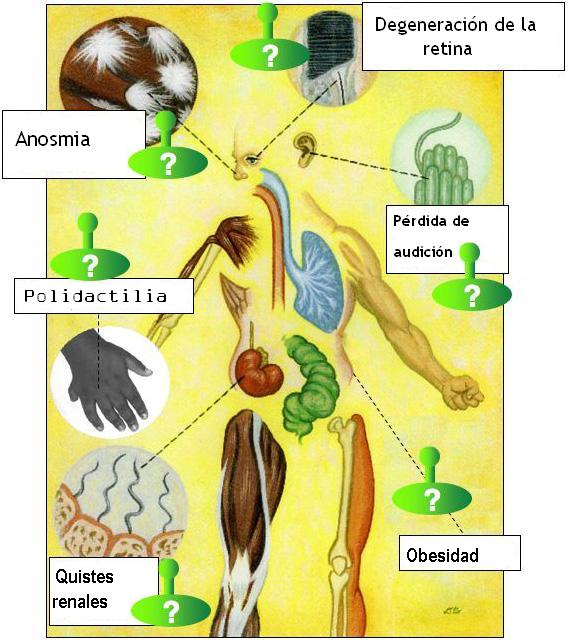
Aviso médico El síndrome de Bardet-Biedl es una enfermedad genética rara de tipo ciliopático que produce efectos muy diversos en los sistemas orgánicos (multisistémica). Principalmente se caracteriza por manifestaciones de obesidad, retinitis pigmentosa, polidactilia, retraso mental, hipogonadismo e insuficiencia renal en algunos casos.[1]
Contenido
Historia y origen del epónimo
El primer caso conocido fue descrito por Zachariah Laurence y Robert Moon en 1886 en el Ophthalmic Hospital de South London.
Clasificación
El sinónimo síndrome de Laurence-Moon-Biedl-Bardet ya no se considera válido, habiéndose dividido en dos entidades nosológicas distintas, puesto que los pacientes de Laurence y Moon presentaban paraplejía pero no polidactilia ni obsesidad. No obstante, existen discrepancias y algunos autores consideran que ambas afecciones podrían no ser diferentess.[2]
Formas del síndrome de Bardet-Biedl[3] Forma Símbolo Gen Locus Otras afecciones del locus Síndrome de Bardet-Biedl1 BBS1 BBS1 11q13 Síndrome de Bardet-Biedl2 BBS2 BBS2 16q21 Síndrome de Bardet-Biedl3 BBS3 ARL6 3p12-q13 Síndrome de Bardet-Biedl4 BBS4 BBS4 15q22.3-q23 Síndrome de Bardet-Bied5 BBS5 2q31 Síndrome de Bardet-Biedl6 BBS6 MKK6 20p12 Síndrome de McKusick-Kaufman Síndrome de Bardet-Biedl7 BBS7 BBS7 4q27 Síndrome de Bardet-Biedl8 BBS8 TTC8 14q32.11 Síndrome de Bardet-Biedl9 BBS9 7p14 Síndrome de Bardet-Biedl10 BBS10 12q Síndrome de Bardet-Biedl11 BBS11 9q31.1 Síndrome de Bardet-Biedl12 BBS12 4q27 Síndrome de Bardet-Biedl13 BBS13 MKS1 17q23 Síndrome de Meckel-Gruber 1 Síndrome de Bardet-Biedl14 BBS14 CEP290 12q21.3 Síndrome de Meckel-Gruber 4 Epidemiología
El síndrome de Bardet Biedl es una enfermedad rara. Su prevalencia varía en gran medida. Croft y Swift determinaron en 1990 esta tasa para la población norteamericana en 1/100 000.[4] Estudios más recientes sitúan la prevalencia en europa en el rango de 1/125 000 a 1/175 000[5] En poblaciones aisladas o con mayor consanguineidad, la prevalencia puede ser mayor. Por ejemplo, entre los beduinos de Kuwait se estima en 1/13 500,[6] y en la isla de Newfoundland, en Terranova es de 1/17 000, posiblemente debido a un efecto fundador.[7]
Síntomas
- Ojos: retinosis pigmentaria, agudeza visual pobre, déficit visual y/o ceguera producido por problemas en el mecanismo de transporte de los fotorreceptores de la retina.[8]
- Nariz: pérdida o reducción del sentido del olfato (anosmia). Algunos pacientes refieren un sentido del olfato más agudo de lo normal.[9]
- pies y manos: polidactilia (dedos extranumerarios) o sindactilia (fusión de dedos).
- Sistema cardiovascular: hipertrofia del tabique interventricular y del ventrículo izquierdo y cardiomiopatía dilatada.
- Aparato digestivo: fibrosis.
- sistema genitourinario: hipogonadismo, fallo renal, anomalías del seno urogenital (cloaca persistente), uretra ectópica, útero doble, vagina septada, e hipoplasia del útero, ovarios, y trompas de Falopio.
- Crecimiento y desarrollo: retraso mental y de crecimiento.
- Conducta y capacidades: se han identificado varios problemas de socialización e interacción social relacionados con el síndrome de Bardet Biedl. Algunos autores se refieren a ellos como una forma de autismo moderada.
- Termosensibilidad y mecanosensibilidad. Un estudio realizado en 2007 determina que las alteraciones genéticas en las proteínas del complejo multiproteico llamado BBSoma producen defectos en la función de los sensores periféricos en la especie humana.[10]
- Otros síntomas: obesidad, probablemente relacionado con un descenso en la función sensorial que en condiciones normales indica saciedad.
Patofisiología
Los mecanismos bioquímicos específicos que conducen al síndrome de Bardet-Biedl siguen siendo un misterio. Hasta el momento, 12 genes se han visto involucrados:(BBS1, BBS2, BBS3, BBS4, BBS5, BBS6, BBS7, BBS8, BBS9, BBS10, BBS11, BBS12). Estos genes que se encuentran mutados en los pacientes con esta enfermedad se han logrado clonar[cita requerida]. Las proteínas codificadas por los 'genes BBS, llamadas proteínas BBS, están localizadas en la porción basal y ciliar de la célula.[11]
Usando al gusano C. elegans como modelo de investigacion, los biólogos han descubierto que estas proteínas están involucraddas en un proceso llamado transporte intraflagelar (IFT) un sistema de transporte bidireccional dentro de los cilios a lo largo del eje longitudinal, siendo esencial para la formación y mantención de los cilios.[12] Recientes análisis bioquímicos de las proteínas BBS en humanos revelaron que las proteínas son ensamblada en múltiples complejos protéicos, llamados BBSoma. Se propone que el BBSoma es el responsable del transporte de vesículas intracelulares hasta la base del cilio jugando un rol importante en su función. Dado que las anomalías en el funcionamiento de los cilios son conocidos y coinciden con la sintomatología que produce el Síndrome de Bardet-Biedi, ahora es ampliamente aceptado que los genes mutados en el BBS afectan las funciones normales de los cilios.[cita requerida]
Herencia
El síndrome es familiar y se transmite como autosómico recesivo. El locus del cromosoma 3 aparece enlazado con la polidactilia de los cuatro miembros, mientras el cromosoma 15 se asocia con obesidad mórbida y es mayormente confinada a manos, y el cromosoma 16 representa la forma "plana".
Tratamiento
El tratamiento es principalmente sintomático, tratando cada una de las complicaciones que aparecen durante la evolución de la enfermedad.
Referencias
- ↑ Beales P, Elcioglu N, Woolf A, Parker D, Flinter F (01 junio 1999). «New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey». J. Med. Genet. 36 (6): pp. 437–46. PMID 10874630. http://mlmorris.com/lmbbs/437.htm.
- ↑ Moore S, Green J, Fan Y et al. (2005). «Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study». Am. J. Med. GenetARRAY 132 (4): pp. 352–60. doi:. PMID 15637713.
- ↑ «OMIM Bardet-Biedl Syndrome» (en inglés). Consultado el 24 de mayo de 2009.
- ↑ Croft, JB; Swift, M (mayo 1990). «Obesity, hypertension, and renal disease in relatives of Bardet-Biedl syndrome sibs» (en inglés). Am J Med Genet. 36 (1): pp. 37-42. PMID 2333905 PMID: 2333905.
- ↑ Rooryck, C; Lacombe, D (diciembre 2008). «Le syndrome de Bardet-Biedl» (en francés). Ann Endocrinol (Paris) 69 (6): pp. 463-71. doi:. PMID 19019343.
- ↑ Farag, TI; Teebi AS (diciembre 1989). «High incidence of Bardet Biedl syndrome among the Bedouin» (en inglés). Clin Genet 36 (6): pp. 463-4. PMID 2591073.
- ↑ Green, JS; Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, Heath O, McManamon PJ, O'Leary E, Pryse-Phillips W. (octubre 1989). «The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome» (en inglés). N Eng J Med 321 (15): pp. 1002-9. PMID 2779627.
- ↑ Abd-El-Barr, MM; Sykoudis K, Andrabi S, Eichers ER, Pennesi ME, Tan PL, Wilson JH, Katsanis N, Lupski JR, Wu SM. (2007-12). «Impaired photoreceptor protein transport and synaptic transmission in a mouse model of Bardet-Biedl syndrome». Vision Res. 47. PMID: 18022666.
- ↑ Downer, Joanna (13 de septiembre de 2004). «That Stinks: People with Rare Obesity Syndrome Can't Sense Odors». The JHU Gazette. Johns Hopkins University. Consultado el 14 de julio de 2008.
- ↑ Tan PL, Barr T, Inglis PN, et al. (2007). «Loss of Bardet Biedl syndrome proteins causes defects in peripheral sensory innervation and function». Proc. Natl. Acad. Sci. U.S.A. 104 (44): pp. 17524–9. doi:. PMID 17959775. PMC 2077289. http://www.pnas.org/content/104/44/17524. }}
- ↑ Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, Kim JC, Ross AJ, Eichers ER, Teslovich TM, Mah AK, Johnsen RC, Cavender JC, Lewis RA, Leroux MR, Beales PL, Katsanis N (October 2003). «Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome». Nature 425 (6958): pp. 628–33. doi:. PMID 14520415.
- ↑ Blacque OE, Reardon MJ, Li C, McCarthy J, Mahjoub MR, Ansley SJ, Badano JL, Mah AK, Beales PL, Davidson WS, Johnsen RC, Audeh M, Plasterk RH, Baillie DL, Katsanis N, Quarmby LM, Wicks SR, Leroux MR. (2004). «Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport.». Genes Dev. 18: pp. 1630–42. doi:. PMID 15231740.
Categoría:- Enfermedades hereditarias
Wikimedia foundation. 2010.