- ATP sintasa
-
El complejo ATP sintasa (EC 3.6.3.14 ) o complejo ATP sintetasa o complejo V o FoF1-ATP sintasa (F = factor de acoplamiento, en inglés coupling factor) es una enzima situada en la cara interna de la membrana interna de las mitocondrias y de la membrana de los tilacoides de los cloroplastos encargada de sintetizar ATP a partir de ADP y un grupo fosfato y la energía suministrada por un flujo de protones (H+). Responde a la síntesis de ATP según la hipótesis quimiosmótica de Mitchell. La síntesis de ATP gracias a este enzima se denomina fosforilación oxidativa (mitocondrias) y fotofosforilación (cloroplastos).
La ATP sintasa se puede imaginar como un motor molecular que produce una gran cantidad de ATP cuando los protones fluyen a través de ella. La tasa de síntesis es grande, el organismo humano en fase de reposo puede formar unas 1021 moléculas de ATP por segundo.[1]
Mediante experimentos in vitro se ha demostrado que la ATP sintasa actúa de forma independiente respecto a la cadena de transporte de electrones, la adición de un ácido débil (por ejemplo ácido acético) a una suspensión de mitocondrias aisladas es suficiente para inducir la biosíntesis de ATP in vitro.[2]
Estructura y funciones de las unidades
La ATP sintasa tiene un diámetro de 10 nm, y es el complejo más pequeño identificado hasta ahora. Trabaja con un grado de efectividad cerca al 100 por ciento.
Esta enzima está formada por dos principales complejos. Una anclada a la membrana mitocondrial interna o al tilacoide llamada F0 (CF0 en caso de los tilacoides) y otra que sobresale por la cara interna de la estructura llamada F1 (CF1 en caso de los tilacoides).
El componente F0 es el motor impulsado por protones. Es conocida como la fracción sensible a la oligomicina está formada por las subunidades a, b2 y c10-14. Las subunidades c forman el “anillo c”, que rota en sentido horario en respuesta al flujo de protones por el complejo. Las dos proteínas b inmovilizan el segundo complejo F1, que está orientada hacia la matriz mitocondrial. Por interacciones electrostáticas, se asocia a F1 a Fo.
F1 está formada por las subunidades α3, β3, γ, δ y ε. La parte principal del complejo F1 está formado por tres diemeres αβ, esta unidad tiene forma de hexamero. La actividad catalitítica de este hexamero está localizada en las subunidades β. Las subunidades γ y ε están unidas al anillo c, y giran con él. Cada rotación de 120º de la subunidad γ induce la aparición de cambios de conformación en los centros catalíticos de las unidads β del los dímeres αβ, provocando la alteración de los centros de fijación de los nuceótidos situado en β. El hexamero α3 y β3 finalmente libera el ATP.
La subunidad Fo consiste de once subunidades diferentes a1 y c10, estas subunidades forman un canal de protones central y finalmente la subunidad b2OSCP1 (Oligomycin Sensitiviy Conferring Protein) enlazan las unidades F1 y Fo.[2]
Hipótesis quimiosmótica
La hipótesis quimiosmótica también conocida como la hipótesis quimiosmótica de Mitchell, explica la manera en que la energía libre que se genera por la actividad de cadena de transporte de electrones se emplea para producir ATP a partir de la reacción ADP + Pi.
Las coenzimas reducidas procedentes del ciclo de Krebs (NADH + H+ y FADH2) atraviesan una serie de complejos enzimáticos (cadena de transporte de electrones) que producen en ellas sucesivas reacciones de oxidación-reducción cuyo objetivo es la utilización del flujo de electrones para bombear protones al espacio intermembrana de la mitocondria o cloroplasto, creando un gradiente electroquímico un gradiente de pH (gradiente de concentración) el y de un desequilibrio de carga (gradiente electroquímico) en la membrana mitocondrial interna. Las ATP sintase forman ATP utilizando la energía libre procedente del gradiente de protones, utilizando el hecho de que los protones tienden a volver al interior del orgánulo, e sólo se pueden hacer a través de la ATP sintasa, por el hecho de que la membrana interna es impermeable a los protones.
Cuando los protones son bombeados fuera de la matriz, el espacio intermembrana se convierte en más ácido y con mayor carga positiva que en la matriz. La matriz es básica y con una carga negativa. Peter Denis Mitchell (1920-1992) utilizó el término “quimiosmótico” para describir las reacciones enzimáticas en las que intervienen, simultáneamente, una reacción química y un proceso de transporte.
Mecanismo de la síntesis de ATP
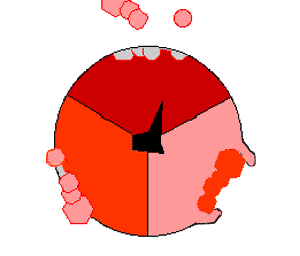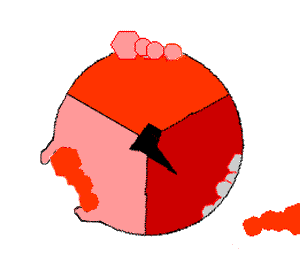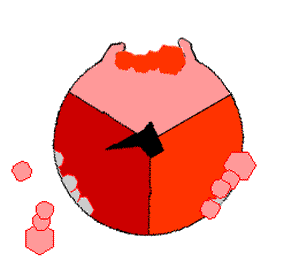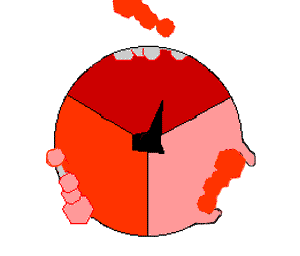
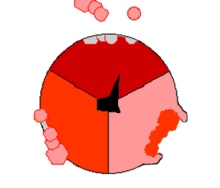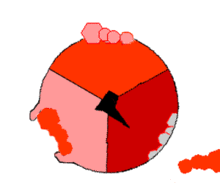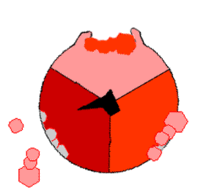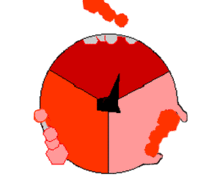 ATPsyntasens mekanisme. ATP en rojo, ADP y fosfato en rosado y la propiedad γ rodando en negro.
ATPsyntasens mekanisme. ATP en rojo, ADP y fosfato en rosado y la propiedad γ rodando en negro.
La síntesis de ATP se escribe algunas veces como:
ADP + Pi + nH+p → ATP + H2O + nH+P
F1 cataliza la síntesis que es fuertemente exergonica de ATP a partir de Pi y ADP. Mecánicamente se impulsa la reacción catalítica con la fuerza protomotriz del gradiente de protones a través de la membrana mitocondrial causando el movimiento de giro del anillo c, γ está unida al anillo c, provocandole movimientos de rotación. Cada rotación de 120º de la subunidad γ induce la apareción de cambios de conformación en los centros catalíticos de las unidades β del los dímeres αβ, de forma que los centros de fijación de nucleótidos van alternando entre tres estados:
Estado O = estado abierto, L = unión libre y T= unión tensa (en inglés, tight).
Aunque la composición de aminoácidos de las tres subunidades β es idéntica, sus conformaciónes difieren en parte por la asociación a al subunidad γ.
Los dímeros αβ son asimétricos, cada uno de ellos presenta una conformación diferente en cada estado. Las tres subunidades β interaccionan de tal modo que, cuando una adopta la conformación O, otra ha de adoptar la conformación L y la del otro una conformación T. La conformación T posee mayor afinidad para ATP que para ADP + Pi y disminuye con ello la constante de velocidad de la reacción en valores cercano a uno; es decir, substrato y producto se encuentran en condiciones estándar, cerca de la equimolaridad.
La síntesis de ATP se inicia en el estado L con la unión de ADP y Pi. El siguiente estado es la conformación T que sigue la condensación del ADP y Pi a ATP con la formación de un enlace fosfodiéster. Finalmente, el estado O deja libre el producto ATP, y vuelve nuevamente al estado L iniciando nuevamente la siguiente ronda de síntesis. Por lo tanto, una rotación completa de la subunidad γ provoca que cada subunidad β se cicle a través de sus tres conformaciones posibles y en cada rotación se sintetizan y se liberan de la superficie del enzima tres moléculas de ATP. La interconvención conducida por protones, direccional y cíclica, de los estados O, L y T, permite una producción continua. Este mecanismo se conoce como mecanismo de cambio de la fijación.[3]
El paso dependiente de energía no es la síntesis de ATP sino su liberación de un lugar de unión compacta. Esta liberación se produce por la rotación de γ que requiere energía, que impulsa los cambios conformacionales de los dímeres αβ. Está liberación se produce simultáneamente con la unión del ADP y el Pi, que se habían unido previamente, se unen a un lugar T para experimentar una conversación espontánea a ATP, mientras que el lugar O, del que se liberó el ATP, une otro ADP y Pi para empezar de nuevo el proceso.[4]
Historia
En 1994, la publicación, por parte del grupo de John Walker, de la estuctura cristalina del componente F1 de 371 Kda del complejo V aclaró el mecanismo que medía el paso de protones a través del complejo FoF1 e impulsa la síntesis de ATP. Todo esto apoyó el mecanismo propuesto por Paul Boyer varios años antes, el mecanismo de catálisis rotacional, en el que la rotación de la subunidad γ produce cambios conformacionales de los dímeres αβ.
La determinación estructural del complejo FoF1 y el mecanismo de la síntesis de ATP condujo al reconocimiento de Walker y Boyer como ganadores del Premio Nobel de Química de 1997.
Acoplamiento del transporte electrónico y la síntesis de ATP
El gradiente electroquímico acopla el ritmo de la cadena de transporte electrónico con el ritmo de la síntesis de ATP. Debido a que el flujo electrónico necesita el bombeo de protones, el flujo electrónico no puede producirse más rápidamente que la utilización de los protones para síntesis de ATP (fosforilación oxidativa acoplada), significando en una relación estrechamente acoplada entre la oxidación y la fosforilación.
Esto conlleva a que los sustratos se oxidan, los electrones se transportan y el oxígeno se consumó tan sólo cuando se requiere la síntesis de ATP. Por lo tanto, las mitocondrias en reposo consumen oxígeno a una velocidad lenta, pero que puede incrementar enormemente en la presencia de ADP. El ADP es captado por las mitocondrias y estimula la ATP sintasa, que disminuye el gradiente de protones. Entonces aumenta la respiración, puesto que son estimuladas las bombas de protones para restablecer el gradiente. Por tanto se puede resumir diciendo que el “control respiratorio” es la dependencia de captación de oxígeno por las mitocondrias según la disponibilidad de ATP.[5]
Desacoplamiento
Desacopladores sintéticos
Se trata de un fenómeno donde los protones retornan a la matriz sin pasar por la ATP sintasa, lo que implica la no generación de ATP. El desacoplamiento es provocado por compuestos químicos, conocidos como desacopladores o como ionóforos protónicos. Los desacoplantes suelen ser compuestos hidrófobicos (bases o ácidos débiles), con un pKa cercano al pH 7, captan protones rapidamente en el espacio intermembrana. Su liposolubilidad les permite difundir a través de la membrana mitocondrial interna transportando los protones liberándolos en el lado de la matriz. La entrada rápida de protones disipa el gradiente de potencial electroquímico, por tanto la ATP sintasa es incapaz de sintetizar ATP, implicando a que la membrana interna pierde su integridad estructural y por tanto las mitocondrias también.[5] En este caso se dice que son porosas (leaky).
El desacoplamiento de la fosforilación oxidativa hace desaparecer el gradiente de protones sin intervención de la ATP sintasa. De este modo se prosigue el transporte de electrones a ritmo rápido, el sistema hace un intento inefectivo de restaurar el gradiente de protones oxidando más combustible y bombeando más protones hacia el exterior de las mitocondrias. La energía producida por el transporte de electrones se descarga en forma de calor en vez de utilizarse para la síntesis de ATP.
El 2,4-dinitrofenol (DNP) es una de la biomoléculas que tiene una función desacopladora, es liposoluble y puede atravesar la membrana siendo capaz de captar protones en un medio ácido. El DNP se recomendó en los Estados Unidos como fármaco adelgazante, basado en el principio de que la disminución de ATP y el aumento de del transporte electrónico estimulaba la oxidación del combustible. Sin embargo, se produjeron varias personas fallecieron como consecuencia de su uso. Aumenta radicalmente el consumo de oxígeno y los combustibles metabólicos, y casi toda la energía metabólica se pierde en forma de calor. Las células mueren tanto a causa del exeso de temperatura como de falta de ATP.[2]
El ácido acetilsalicílico desacopla la fosforilación oxidativaa dosis elevadas. Esto explica la fiebre que acompaña a las sobredosis tóxicas de estos fármacos.[6] Otros desacopladores son algunos conservantes y los antimicrobianos (por ej. pentaclorofenol y p-cresol).
Proteínas desacopladodras (UCP)
Ocurre en la membrana mitocondrial interior de los mamíferos. Estas proteínas permiten que los protones reingresen en la matriz mitocondrial sin que se capure energía en forma de ATP.[6]
La primera de estas UCP (uncoupling proteins) que se descubrió fue la proteína de desacoplamiento 1 (UCP1), llamada también termogenina y que se encuentra de modo exclusivo en el tejido adiposo pardo. El tejido adiposo pardo es abundante en el recién nacido y en algunos mamíferos adultos. Además, al parececer su color marrón se debe a su elevado contenido en mitocondrias. La única función. La UCP1 se encarga de activar la oxidación de los ácidos grasos y la producción de calor en el tejido adiposo pardo, proporcionando calor corporal durante el estrés por frío en los animales jóvenes y algunos adultos así como durante la hibernación. Ello se consigue desacoplando el gradiente de protones y generando así calor (termogénesis) en lugar de ATP.
El genoma humano expresa otras cuatro proteínas desacoplantes: UCP2, UCP3, UCP4 y UCP5. El UCP1 es exclusiva del tejido adiposo pardo, UCP2 se expresa de modo ubicuo, UPC3 se expresa principalmente en el músculo esquelético, y el UCP4 y UCP5 se expresa en el cerebro. Con la expresión de UCP1, en se conocen bien las funciones fisiológicas de estas proteínas, sin embargo, podrían tener gran importancia para conocer mejor trastornos y enfermedades como la diabetes, la obesidad, el cáncer, las enfermedades del tiroides y el envejecimiento. No obstante el mecanismo real aún se desconoce. Asimismo, el sistema de las UCP también puede ser importante en la regulación del potencial de membrana.[2]
Normalmente las mitocondrias humanas sufren un bajo nivel de escape de protones a través de la membrana mitocondrial intrena, y de esta forma, las mitocondrias normalmente se encuentran desacopladas. Las mitocondrias pueden desacoplarse parcialmente
Las reacciones dependientes de la luz tienen lugar en las secciones de la membrana de los cloroplastos y tilacoides, que ofrecen una gran superficie para absorber energía de la luz.
La principal función de estas reacciones consiste en proveer una fuente de ATP y NADP reducido que se usan para reducir CO2 en las reacciones independientes de la luz.
El ATP se forma en el último paso cuando los iones H + son bombeados desde la luz del tilacoide hasta el estroma del cloroplasto a través de la enzima ATP sintasa. Esto transforma a las moléculas de APD en moléculas de ATP.
Inhibidores de la ATP sintasa
La oligomicina[6] se fija en el tallo de la ATP sintasa, inhibiendo el canal de protones, por tanto previniendo el reingreso de protones en la matriz mitocondrial. Como los gradientes de pH y eléctricos no se pueden disipar en presencia de la oligomicina, el transportador de protones se detiene por la dificultad para el bombeo de más protones contra los gradiententes muy inclinados. Causa una acumulación de protones en el espacio intermembrana.[2]
Transporte de ATP y ADP
La fuerza protón-motriz también sirve para impulsar varios procesos de transporte esenciales para la fosforilación oxidativa, aunque el papel principal del gradiente de protones en las mitocondrias es suministrar energía para la síntesis de ATP. La membrana mitocondrial interior necesita transportadores especializados para el ADP y Pi desde el citosol hacia el interior de la mitocondria, al sitio donde se puede resíntetizar a ATP, ya que la membrana mitocondrial interna es generalmente impermeable a las moléculas con carga. Por tanto hay dos sistemas específicos que transportan ADP y Pi a la matriz y ATP hacia el citosol externo. Un transportador de nucleótidos de adenina translocasa y fosfato translocasa son los dos sitemas que intervienen.[4] La nucleótido de adenina translocasa une ADP y es por tanto la encargada en transportar una molécula de ADP desde el citosol hacia el interior de la mitocondria, a la vez que exporta un ATP desde la matriz de nuevo hacia el citosol. Fosfato translocasa transporta en Pi junto con un protón a la matriz.[6]
La toxina de las plantas atractilósido inhibe de manera poderosa al transportador, lo que resulta en un agotamiento de la reserva de ADP intramitrocondrial e interrupción de la producción de ATP[6]
Deficiencia de la ATP sintasa
Los trastornos de la fosforilación oxidativa mitocondrial (OXPHOS) causan un grupo altamente diverso de enfermedades que afectan principalmente a los tejidos que exigen energía, tales como el sistema nervioso, el músculo esquelético y el corazón. Los defectos genéticos generan mutaciones en el DNA mitocondrial (mtDNA) como en los genes nucleares. Mutaciones en el DNA nuclear se involucran con más frecuencia en enfermedades pediátricas de deficiencia mitocondrial que las mutaciones del mtDNA. Los trastornos mendelianos del OXPHOS pueden afectar a todos los complejos de la cadena respiratoria, sobre todo del complejo I y IV, mientras que los trastornos de la ATP sintasa se han reportado con menos frecuencia.
La disfunción aislada de la ATP sintasa puede ser causada tanto por mutaciones del DNA mitocondrial como por defectos genéticos de origen nuclear. Se han descrito varias mutaciones en el gen ATP6 en el mtDNA que alteran la función del canal de protones y provocan pérdida de actividad de síntesis de ATP, mientras que mantiene la actividad hidrolítica. Los trastornos transmitidos por vía materna se producen con frecuencia y suelen afectar al sistema nervioso periférico. Los pacientes presentan una neuropatía, ataxia y retinitis pigmentosa (NARP), síndrome de Leigh heredado por vía materna (MILS) o necrosis estriatal bilateral.
El origen nuclear de la deficiencia de la ATP sintasa se demostró por primera vez en 1999 en un niño con grave acidosis láctica, cardiomiopatía y hepatomegalia. Hasta ahora, sólo unos pocos han sido reportados sólo con deficiencia de la ATP sintasa debido a defectos genéticos en el núcleo. Se caracterizan por un contenido marcadamente disminuido de manera estructural y por un funcionamiento normal de la enzima. La causa principal parece ser un defecto en la fase inicial del ensamblado de la enzima, que incluye la formación de la parte catalítica de F1 de la enzima.[7]
Las síntomas sobre cada uno de los complejos defectos no se pueden describir claramente, suele suceder que un mismo individuo presenta defectos en más de un complejo.
Véase también
Referencias
- ↑ *Werner, Múller-Esterl (2008). «37». Bioquímica, Fundamento para Medicina y Ciencias de la Vida (1 edición). Editorial Reverté. pp. 518. ISBN 978-84-291-7393-2.
- ↑ a b c d e * Smith, Marks, Lieberman. «21». Bioquímica básica de Marks, un enfoque clínico (segunda edición). Lippincott Williams & Wilkins. pp. 325-330. ISBN 0-7817-2145-8.
- ↑ * Cox, Nelson (2004). «19». Lehninger (quarta edición). Palgrave Macmillan. pp. 704-723. ISBN 0-7167-4339-6.
- ↑ a b * Mathews, Van Holde, Attern. «15». Bioquímica (tercera edición). Pearson Educación, S.A.. pp. 600-615. ISBN 84-7829-053-Z.
- ↑ a b * Smith, Marks, Lieberman. «21». Bioquímica básica de Marks, un enfoque clínico (segunda edición). Lippincott Williams & Wilkins. pp. 325-330. ISBN 0-7817-2145-8.
- ↑ a b c d e * Champe, Harvey, Ferrier, Pamela C., Richard A., Denise R. (2005). «6». Bioquímica (tercera edición). MCGRAW HILL. pp. 88-91. ISBN 970-10-5377-X.
- ↑ * W. Sperla, P. Ješinab, J. Zemanc,. «Deficiency of mitochondrial ATP synthase of nuclear genetic origin» (en inglés). Consultado el 08.08 de 2006.
Enlaces externos
- Animación que muestra cómo trabaja la ATP sintasa
- Harvard Multimedia Production Site - Videos - Animación sobre la ATP sintasa
Categorías:- Enzimas
- Mitocondria
Wikimedia foundation. 2010.