- Genoma humano
-
El genoma humano es el genoma del Homo sapiens, es decir, la secuencia de ADN contenida en 23 pares de cromosomas en el núcleo de cada célula humana diploide.
De los 23 pares, 22 son cromosomas autosómicos y un par determinante del sexo (dos cromosomas X en mujeres y uno X y uno Y en hombres). El genoma haploide (es decir, con una sola representación de cada par) tiene una longitud total aproximada de 3200 millones de pares de bases de ADN (3200 Mb) que contienen unos 20.000-25.000 genes[1] (las estimaciones más recientes apuntan a unos 20.500). De las 3200 Mb unas 2950 Mb corresponden a eucromatina y unas 250 Mb a heterocromatina. El Proyecto Genoma Humano produjo una secuencia de referencia del genoma humano eucromático, usado en todo el mundo en las ciencias biomédicas.
La secuencia de ADN que conforma el genoma humano contiene codificada la información necesaria para la expresión, altamente coordinada y adaptable al ambiente, del proteoma humano, es decir, del conjunto de las proteínas del ser humano. Las proteínas, y no el ADN, son las principales biomoléculas efectoras; poseen funciones estructurales, enzimáticas, metabólicas, reguladoras, señalizadoras..., organizándose en enormes redes funcionales de interacciones. En definitiva, el proteoma fundamenta la particular morfología y funcionalidad de cada célula. Asimismo, la organización estructural y funcional de las distintas células conforma cada tejido y cada órgano, y, finalmente, el organismo vivo en su conjunto. Así, el genoma humano contiene la información básica necesaria para el desarrollo físico de un ser humano completo.
El genoma humano presenta una densidad de genes muy inferior a la que inicialmente se había predicho, con sólo en torno al 1,5%[2] de su longitud compuesta por exones codificantes de proteínas. Un 70% está compuesto por ADN extragénico y un 30 % por secuencias relacionadas con genes. Del total de ADN extragénico, aproximadamente un 70% corresponde a repeticiones dispersas, de manera que, más o menos, la mitad del genoma humano corresponde a secuencias repetitivas de ADN. Por su parte, del total de ADN relacionado con genes se estima que el 95% corresponde a ADN no codificante: pseudogenes, fragmentos de genes, intrones o secuencias UTR, entre otros.
Contenido en genes y tamaño del genoma de varios organismos[3] Especie Tamaño del
genoma (Mb)Número
de genesMycoplasma genitalium 0,58 500 Streptococcus pneumoniae 2,2 2300 Escherichia coli 4,6 4.400 Saccharomyces cerevisiae 12 5.800 Caenorhabditis elegans 97 19.000 Arabidopsis thaliana 125 25.500 Drosophila melanogaster (mosca) 180 13.700 Oryza sativa (arroz) 466 45-55.000 Mus musculus (ratón) 2500 29.000 Homo sapiens (ser humano) 2900 27.000 Contenido
Componentes
Cromosomas
El genoma humano (como el de cualquier organismo eucariota) está formado por cromosomas, que son largas secuencias continuas de ADN altamente organizadas espacialmente (con ayuda de proteínas histónicas y no histónicas) para adoptar una forma ultracondensada en metafase. Son observables con microscopía óptica convencional o de fluorescencia mediante técnicas de citogenética y se ordenan formando un cariotipo.
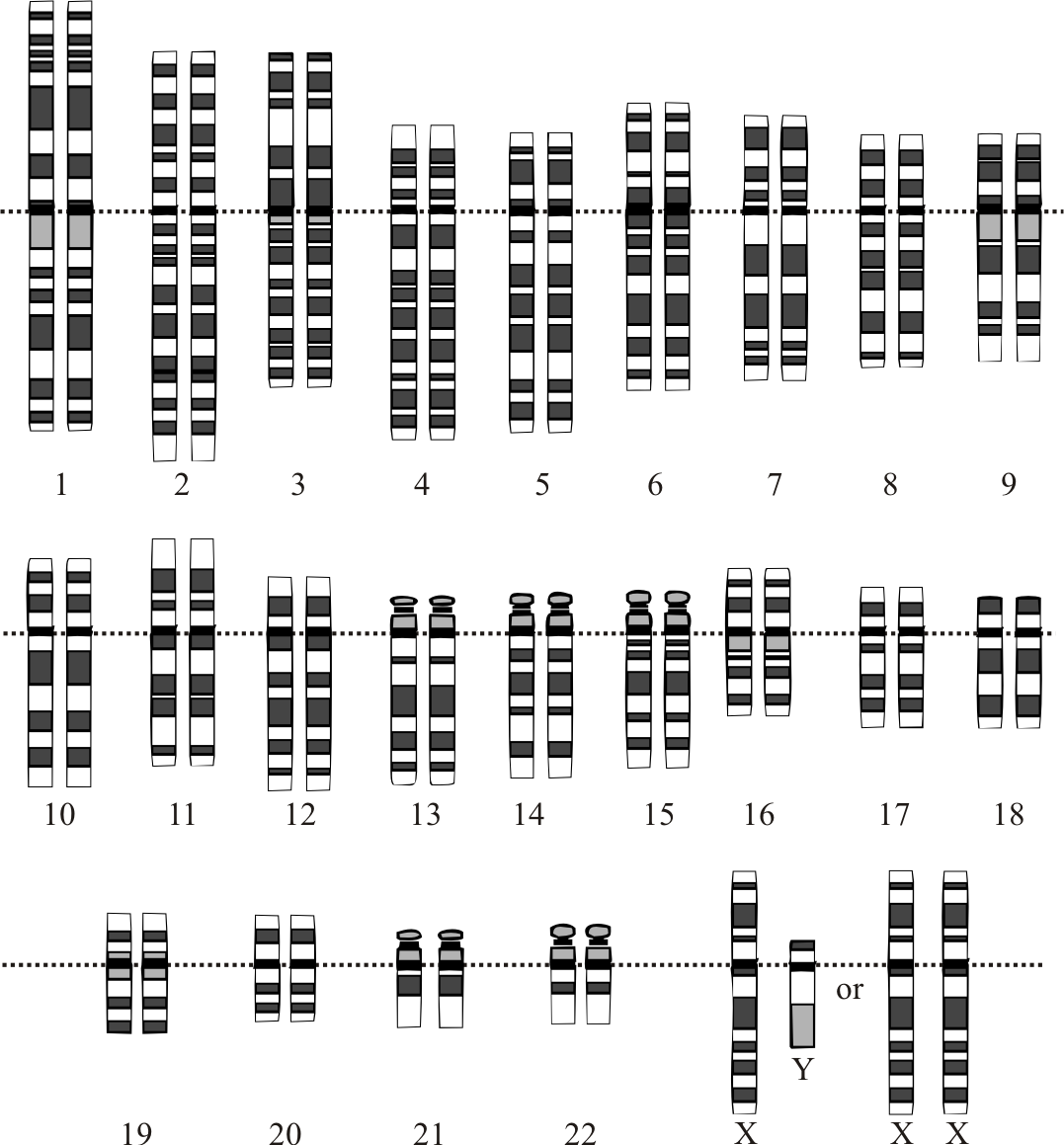
El cariotipo humano normal contiene un total de 23 pares de cromosomas distintos: 22 pares de autosomas más 1 par de cromosomas sexuales que determinan el sexo del individuo. Los cromosomas 1-22 fueron numerados en orden decreciente de tamaño en base al cariotipo. Sin embargo, posteriormente pudo comprobarse que el cromosoma 22 es en realidad mayor que el 21.
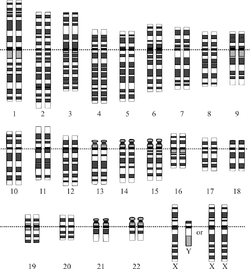 Representación gráfica del cariotipo humano normal.(Imagen 1).
Representación gráfica del cariotipo humano normal.(Imagen 1).
Las células somáticas de un organismo poseen en su núcleo un total de 46 cromosomas (23 pares): una dotación de 22 autosomas procedentes de cada progenitor y un par de cromosomas sexuales, un cromosoma X de la madre y un X o un Y del padre. (Ver imagen 1). Los gametos -óvulos y espermatozoides- poseen una dotación haploide de 23 cromosomas.
Cromosoma Genes Número de bases Bases secuenciadas[4] 1 4.220 247.199.719 224.999.719 2 1.491 242.751.149 237.712.649 3 1.550 199.446.827 194.704.827 4 446 191.263.063 187.297.063 5 609 180.837.866 177.702.766 6 2.281 170.896.993 167.273.993 7 2.135 158.821.424 154.952.424 8 1.106 146.274.826 142.612.826 9 1.920 140.442.298 120.312.298 10 1.793 135.374.737 131.624.737 11 379 134.452.384 131.130.853 12 1.430 132.289.534 130.303.534 13 924 114.127.980 95.559.980 14 1.347 106.360.585 88.290.585 15 921 100.338.915 81.341.915 16 909 88.822.254 78.884.754 17 1.672 78.654.742 77.800.220 18 519 76.117.153 74.656.155 19 1.555 63.806.651 55.785.651 20 1.008 62.435.965 59.505.254 21 578 46.944.323 34.171.998 22 1.092 49.528.953 34.893.953 X (cromosoma sexual) 1.846 154.913.754 151.058.754 Y (cromosoma sexual) 454 57.741.652 25.121.652 Total 32.185 3.079.843.747 2.857.698.560 ADN intragénico
Genes
Un gen es la unidad básica de la herencia, y porta la información genética necesaria para la síntesis de una proteína (genes codificantes) o de un ARN no codificante (genes de ARN). Está formado por una secuencia promotora, que regula su expresión, y una secuencia que se transcribe, compuesta a su vez por: secuencias UTR (regiones flanqueantes no traducidas), necesarias para la traducción y la estabilidad del ARNm, exones (codificantes) e intrones, que son secuencias de ADN no traducidas situadas entre dos exones que serán eliminadas en el procesamiento del ARNm (ayuste).
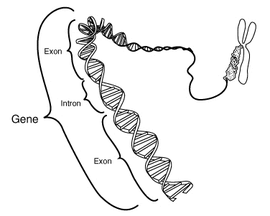 Este diagrama esquemático muestra un gen en relación a su estructura física (doble hélice de ADN) y a un cromosoma (derecha). Los intrones son regiones frecuentemente encontradas en los genes de eucariotas, que se transcriben, pero son eliminadas en el procesamiento del ARN (ayuste) para producir un ARNm formado sólo por exones, encargados de traducir una proteína. Este diagrama es en exceso simplificado ya que muestra un gen compuesto por unos 40 pares de bases cuando en realidad su tamaño medio es de 20.000-30.000 pares de bases).
Este diagrama esquemático muestra un gen en relación a su estructura física (doble hélice de ADN) y a un cromosoma (derecha). Los intrones son regiones frecuentemente encontradas en los genes de eucariotas, que se transcriben, pero son eliminadas en el procesamiento del ARN (ayuste) para producir un ARNm formado sólo por exones, encargados de traducir una proteína. Este diagrama es en exceso simplificado ya que muestra un gen compuesto por unos 40 pares de bases cuando en realidad su tamaño medio es de 20.000-30.000 pares de bases).Actualmente se estima que el genoma humano contiene entre 20.000 y 25.000 genes codificantes de proteínas, estimación muy inferior a las predicciones iniciales que hablaban de unos 100.000 genes o más. Esto implica que el genoma humano tiene menos del doble de genes que organismos eucariotas mucho más simples, como la mosca de la fruta o el nematodo Caenorhabditis elegans. Sin embargo, las células humanas recurren ampliamente al splicing (ayuste) alternativo para producir varias proteínas distintas a partir de un mismo gen, como consecuencia de lo cual el proteoma humano es más amplio que el de otros organismos mucho más simples. En la práctica, el genoma tan sólo porta la información necesaria para una expresión perfectamente coordinada y regulada del conjunto de proteínas que conforman el proteoma, siendo éste el encargado de ejecutar la mayor parte de las funciones celulares.
Con base en los resultados iniciales arrojados por el proyecto ENCODE[5] (acrónimo de ENCyclopedia Of DNA Elements), algunos autores han propuesto redefinir el concepto actual de gen. Las observaciones más recientes hacen difícilmente sostenible la visión tradicional de un gen, como una secuencia formada por las regiones UTRs, los exones y los intrones. Estudios detallados han hallado un número de secuencias de inicio de transcripción por gen muy superior a las estimaciones iniciales, y algunas de estas secuencias se sitúan en regiones muy alejadas de la traducida, por lo que los UTR 5' pueden abarcar secuencias largas dificultando la delimitación del gen. Por otro lado, un mismo transcrito puede dar lugar a ARN maduros totalmente diferentes (ausencia total de solapamiento), debido a una gran utilización del splicing alternativo. De este modo, un mismo transcrito primario puede dar lugar a proteínas de secuencia y funcionalidad muy dispar. En consecuencia, algunos autores han propuesto una nueva definición de gen,:[6] [7] la unión de secuencias genómicas que codifican un conjunto coherente de productos funcionales, potencialmente solapantes. De este modo, se identifican como genes los genes ARN y los conjuntos de secuencias traducidas parcialmente solapantes (se excluyen, así, las secuencias UTR y los intrones, que pasan a ser considerados como "regiones asociadas a genes", junto con los promotores). De acuerdo con esta definición, un mismo transcrito primario que da lugar a dos transcritos secundarios (y dos proteínas) no solapantes debe considerarse en realidad dos genes diferentes, independientemente de que estos presenten un solapamiento total o parcial de sus transcritos primarios.
Las nuevas evidencias aportadas por ENCODE, según las cuales las regiones UTR no son fácilmente delimitables y se extienden largas distancias, obligarían a reidentificar nuevamente los genes que en realidad componen el genoma humano. De acuerdo con la definición tradicional (actualmente vigente), sería necesario identificar como un mismo gen a todos aquellos que muestren un solapamiento parcial (incluyendo las regiones UTR y los intrones), con lo que a la luz de las nuevas observaciones, los genes incluirían múltiples proteínas de secuencia y funcionalidad muy diversa. Colateralmente se reduciría el número de genes que componen el genoma humano. La definición propuesta, en cambio, se fundamenta en el producto funcional del gen, por lo que se mantiene una relación más coherente entre un gen y una función biológica. Como consecuencia, con la adopción de esta nueva definición, el número de genes del genoma humano aumentará significativamente.
Genes de ARN
Además de los genes codificantes de proteínas, el genoma humano contiene varios miles de genes ARN, cuya transcripción reproduce ARN de transferencia (ARNt), ARN ribosómico (ARNr), microARN (miARN), u otros genes ARN no codificantes. Los ARN ribosómico y de transferencia son esenciales en la constitución de los ribosomas y en la traducción de las proteínas. Por su parte, los microADN tienen gran importancia en la regulación de la expresión génica, estimándose que hasta un 20-30% de los genes del genoma humano puede estar regulado por el mecanismo de interferencia por miARN. Hasta el momento se han identificado más de 300 genes de miARN y se estima que pueden existir unos 500-
Distribución de genes
A continuación se muestran algunos valores promedio del genoma humano. Cabe advertir, sin embargo, que la enorme heterogeneidad que presentan estas variables hace poco representativos a los valores promedio, aunque tienen valor orientativo.
La densidad media de genes es de 1 gen cada 100 kb, con un tamaño medio de 20-30 kb, y un número de exones promedio de 7-8 por cada gen, con un tamaño medio de 150 nucleótidos. El tamaño medio de un ARNm es de 1,8-2,2 kb, incluyendo las regiones UTR (regiones no traducidas flanqueantes), siendo la longitud media de la región codificante de 1,4 kb.
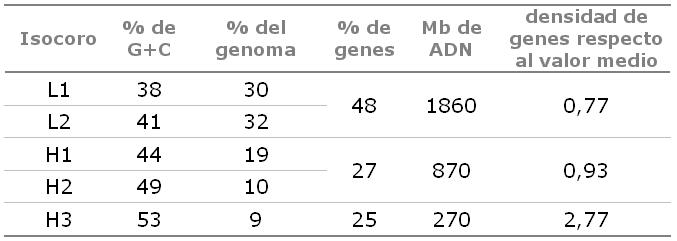
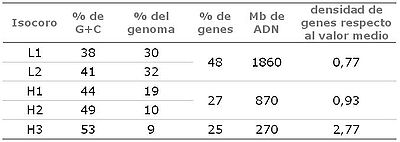 Isocoros. Frecuencia y riqueza en G+C y genes, en el genoma humano.
Isocoros. Frecuencia y riqueza en G+C y genes, en el genoma humano.El genoma humano se caracteriza por presentar una gran heterogeneidad en su secuencia. En particular, la riqueza en bases de guanina (G) y citosina (C) frente a las de adenina (A) y timina (T) se distribuye heterogéneamente, con regiones muy ricas en G+C flanqueadas por regiones muy pobres, siendo el contenido medio de G+C del 41%, menor al teóricamente esperado (50%). Dicha heterogeneidad esta correlacionada con la riqueza en genes, de manera que los genes tienden a concentrarse en las regiones más ricas en G+C. Este hecho era conocido ya desde hace años gracias a la separación mediante centrifugación en gradiente de densidad de regiones ricas en G+C (que recibieron el nombre de isócoros H; del inglés High) y regiones ricas en A+T (isócoros L; del inglés Low).
Secuencias reguladoras
El genoma tiene diversos sistemas de regulación de la expresión génica, basados en la regulación de la unión de factores de transcripción a las secuencias promotoras, en mecanismos de modificación epigenética (metilación del ADN o metilación-acetilación de histonas) o en el control de la accesibilidad a los promotores determinada por el grado de condensación de la cromatina; todos ellos muy interrelacionados. Además hay otros sistemas de regulación a nivel del procesamiento, estabilidad y traducción del ARNm, entre otros. Por lo tanto, la expresión génica está intensamente regulada, lo cual permite desarrollar los múltiples fenotipos que caracterizan los distintos tipos celulares de un organismo eucariota multicelular, al mismo tiempo que dota a la célula de la plasticidad necesaria para adaptarse a un medio cambiante. No obstante, toda la información necesaria para la regulación de la expresión génica, en función del ambiente celular, está codificada en la secuencia de ADN al igual que lo están los genes.
Las secuencias reguladoras son típicamente secuencias cortas presentes en las proximidades o en el interior (frecuentemente en intrones) de los genes. En la actualidad, el conocimiento sistemático de estas secuencias y de cómo actúan en complejas redes de regulación génica, sensibles a señales exógenas, es muy escaso y está comenzando a desarrollarse mediante estudios de genómica comparada, bioinformática y biología de sistemas. La identificación de secuencias reguladoras se basa en parte en la búsqueda de regiones no codificantes evolutivamente conservadas.[8] Por ejemplo, la divergencia evolutiva entre el ratón y el ser humano ocurrió hace 70-90 millones de años.[9] Mediante estudios de genómica comparada, alineando secuencias de ambos genomas pueden identificarse regiones con alto grado de coincidencia, muchas correspondientes a genes y otras a secuencias no codificantes de proteínas pero de gran importancia funcional, dado que han estado sometidas a presión selectiva.
Elementos ultraconservados
Reciben este nombre regiones que han mostrado una constancia evolutiva casi total, mayor incluso que las secuencias codificantes de proteínas, mediante estudios de genómica comparada. Estas secuencias generalmente se solapan con intrones de genes implicados en la regulación de la transcripción o en el desarrollo embrionario y con exones de genes relacionados con el procesamiento del ARN. Su función es generalmente poco conocida, pero probablemente de extrema importancia dado su nivel de conservación evolutiva, tal y como se ha expuesto en el punto anterior.
En la actualidad se han encontrado unos 500 segmentos de un tamaño mayor a 200 pares de bases totalmente conservados (100% de coincidencia) entre los genomas de humano, ratón y rata, y casi totalmente conservados en perro (99%) y pollo (95%).[10]
Pseudogenes
En el genoma humano se han encontrado asimismo unos 19.000 pseudogenes, que son versiones completas o parciales de genes que han acumulado diversas mutaciones y que generalmente no se transcriben. Se clasifican en pseudogenes no procesados (~30%) y pseudogenes procesados (~70%)[11]
- Los pseudogenes no procesados son copias de genes generalmente originadas por duplicación, que no se transcriben por carecer de una secuencia promotora y haber acumulado múltiples mutaciones, algunas de las cuales sin sentido (lo que origina codones de parada prematuros). Se caracterizan por poseer tanto exones como intrones.
- Los pseudogenes procesados, por el contrario, son copias de ARN mensajero retrotranscritas e insertadas en el genoma. En consecuencia carecen de intrones y de secuencia promotora.
ADN intergénico
Como se ha dicho, las regiones intergénicas o extragénicas comprenden la mayor parte de la secuencia del genoma humano, y su función es generalmente desconocida. Buena parte de estas regiones está compuesta por elementos repetitivos, clasificables como repeticiones en tándem o repeticiones dispersas, aunque el resto de la secuencia no responde a un patrón definido y clasificable. Gran parte del ADN intergénico puede ser un artefacto evolutivo sin una función determinada en el genoma actual, por lo que tradicionalmente estas regiones han sido denominadas ADN "basura" (Junk DNA), denominación que incluye también las secuencias intrónicas y pseudogenes. No obstante, esta denominación no es la más acertada dado el papel regulador conocido de muchas de estas secuencias. Además el notable grado de conservación evolutiva de algunas de estas secuencias parece indicar que poseen otras funciones esenciales aún desconocidas o poco conocidas. Por lo tanto, algunos prefieren denominarlo "ADN no codificante" (aunque el llamado "ADN basura" incluye también transposones codificantes) o "ADN repetitivo". Algunas de estas regiones constituyen en realidad genes precursores para la síntesis te microARN (reguladores de la expresión génica y del silenciamiento génico).
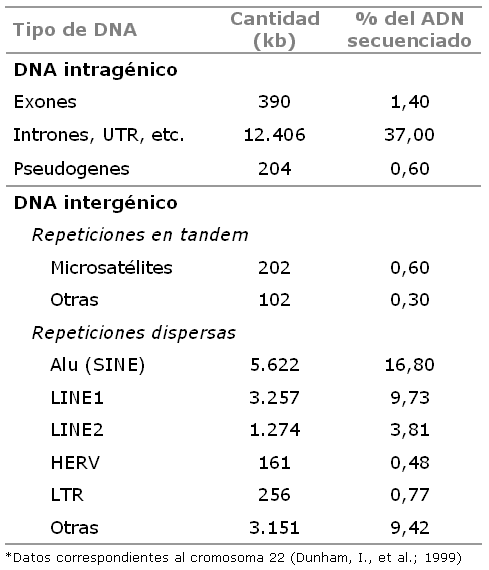
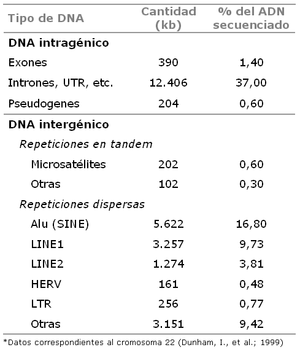 Frecuencia de las diversas regiones intergénicas e intragénicas del cromosoma 22. Adaptado de: Dunham, I., et al., 1999. The DNA sequence of human chromosome 22, Nature 402(6761):489–495.
Frecuencia de las diversas regiones intergénicas e intragénicas del cromosoma 22. Adaptado de: Dunham, I., et al., 1999. The DNA sequence of human chromosome 22, Nature 402(6761):489–495.Estudios recientes enmarcados en el proyecto ENCODE han obtenido resultados sorprendentes, que exigen la reformulación de nuestra visión de la organización y la dinámica del genoma humano. Según estos estudios, el 15% de la secuencia del genoma humano se transcribe a ARN maduros, y hasta el 90% se transcribe al menos a transcritos inmaduros en algún tejido:[7] Así, una gran parte del genoma humano codifica genes de ARN funcionales. Esto es coherente con la tendencia de la literatura científica reciente a asignar una importancia creciente al ARN en la regulación génica. Asimismo, estudios detallados han identificado un número mucho mayor de secuencias de inicio de transcripción por gen, algunas muy alejadas de la región próxima a la traducida. Como consecuencia, actualmente resulta más complicado definir una región del genoma como génica o intergénica, dado que los genes y las secuencias relacionadas con los genes se extienden en las regiones habitualmente consideradas intergénicas.
ADN repetido en tándem
Son repeticiones que se ordenan de manera consecutiva, de modo que secuencias idénticas, o casi, se disponen unas detrás de otras.
Satélites
El conjunto de repeticiones en tándem de tipo satélite comprende un total de 250 Mb del genoma humano. Son secuencias de entre 5 y varios cientos de nucleótidos que se repiten en tándem miles de veces generando regiones repetidas con tamaños que oscilan entre 100 kb (100.000 nucleótidos) hasta varias megabases.
Reciben su nombre de las observaciones iniciales de centrifugaciones en gradiente de densidad del ADN genómico fragmentado, que reportaban una banda principal correspondiente a la mayor parte del genoma y tres bandas satélite de menor densidad. Esto se debe a que las secuencias satélite tienen una riqueza en nuclétidos A+T superior a la media del genoma y en consecuencia son menos densas.
Hay principalmente 6 tipos de repeticiones de ADN satélite[10]
- Satélite 1: secuencia básica de 42 nucleótidos. Situado en los centrómeros de los cromosomas 3 y 4 y el brazo corto de los cromosomas acrocéntricos (en posición distal respecto al cluster codificante de ARNr).
- Satélite 2: la secuencia básica es ATTCCATTCG. Presente en las proximidades de los centrómeros de los cromosomas 2 y 10, y en la constricción secundaria de 1 y 16.
- Satélite 3: la secuencia básica es ATTCC. Presente en la constricción secundaria de los cromosomas 9 e Y, y en posición proximal respecto al cluster de ADNr del brazo corto de los cromosomas acrocéntricos.
- Satélite alfa: secuencia básica de 171 nucleótidos. Forma parte del ADN de los centrómeros cromosómicos.
- Satélite beta: secuencia básica de 68 nucleótidos. Aparece en torno al centrómero en los cromosomas acrocéntricos y en la constricción secundaria del cromosoma 1.
- Satélite gamma: secuencia básica de 220 nucleótidos. Próximo al centrómero de los cromosomas 8 y X.
Minisatélites
Están compuestas por una unidad básica de secuencia de 6-25[10] nucleótidos que se repite en tándem generando secuencias de entre 100 y 20.000 pares de bases. Se estima que el genoma humano contiene unos 30.000 minisatélites.
Diversos estudios han relacionado los minisatélites con procesos de regulación de la expresión génica, como el control del nivel de transcripción, el ayuste (splicing) alternativo o la impronta (imprinting). Asimismo, se han asociado con puntos de fragilidad cromosómica dado que se sitúan próximos a lugares preferentes de rotura cromosómica, translocación genética y recombinación meiótica. Por último, algunos minisatélites humanos (~10%) son hipermutables, presentando una tasa media de mutación entre el 0.5% y el 20% en las células de la línea germinal, siendo así las regiones más inestables del genoma humano conocidas hasta la fecha.
En el genoma humano, aproximadamente el 90% de los minisatélites se sitúan en los telómeros de los cromosomas. La secuencia básica de seis nucleótidos TTAGGG se repite miles de veces en tándem, generando regiones de 5-20 kb que conforman los telómeros.
Algunos minisatélites por su gran inestabilidad presentan una notable variabilidad entre individuos distintos. Se consideran polimorfismos multialélicos, dado que pueden presentarse en un número de repeticiones muy variable, y se denominan VNTR (acrónimo de Variable number tandem repeat). Son marcadores muy utilizados en genética forense, ya que permiten establecer una huella genética característica de cada individuo, y son identificables mediante Southern blot e hibridación.
Microsatélites
Están compuestos por secuencias básicas de 2-4 nucleótidos, cuya repetición en tándem origina frecuentemente secuencias de menos de 150 nucleótidos. Algunos ejemplos importantes son el dinucleótido CA y el trinucleótido CAG.
Los microsatélites son también polimorfismos multialélicos, denominados STR (acrónimo de Short Tandem Repeats) y pueden identificarse mediante PCR, de modo rápido y sencillo. Se estima que el genoma humano contiene unos 200.000 microsatélites, que se distribuyen más o menos homogéneamente, al contrario que los minisatélites, lo que los hace más informativos como marcadores.
ADN repetido disperso
Son secuencias de ADN que se repiten de modo disperso por todo el genoma, constituyendo el 45% del genoma humano. Los elementos cuantitativamente más importantes son los LINEs y SINEs, que se distinguen por el tamaño de la unidad repetida.
Estas secuencias tienen la potencialidad de autopropagarse al transcribirse a una ARNm intermediario, retrotranscribirse e insertarse en otro punto del genoma. Este fenómeno se produce con una baja frecuencia, estimándose que 1 de cada 100-200 neonatos portan una inserción nueva de un Alu o un L1, que pueden resultar patogénicos por mutagénesis insercional, por desregulación de la expresión de genes próximos (por los propios promotores de los SINE y LINE) o por recombinación ilegítima entre dos copias idénticas de distinta localización cromosómica (recombinación intra o intercromosómica), especialmente entre elementos Alu.
Frecuencias y tipos de repeticiones dispersas en el genoma de varios organismos[10] Tipo repetición Homo
sapiensDrosophila
melanogasterCaenorhabditis
elegansArabidopsis
thalianaLINE,SINE 33,4% 0,7% 0,4% 0,5% LTR/HERV 8,1% 1,5% 0% 4,8% Transposones ADN 2,8% 0,7% 5,3% 5,1% Total 44,4% 3,1% 6,5% 10,4% SINE
Acrónimo del inglés Short Interspersed Nuclear Elements (Elementos nucleares dispersos cortos). Son secuencias cortas, generalmente de unos pocos cientos de bases, que aparecen repetidas miles de veces en el genoma humano. Suponen el 13% del genoma humano,[10] un 10% debido exclusivamente a la familia de elementos Alu (característica de primates).
Los elementos Alu son secuencias de 250-280 nucleótidos presentes en 1.500.000[10] de copias dispersas por todo el genoma. Estructuralmente son dímeros casi idénticos, excepto que la segunda unidad contiene un inserto de 32 nucleótidos, siendo mayor que la primera. En cuanto a su secuencia, tienen una considerable riqueza en G+C (56%),[10] por lo que predominan en las bandas R, y ambos monómeros presentan una cola poliA (secuencia de adeninas) vestigio de su origen de ARNm. Además poseen un promotor de la ARN polimerasa III para transcribirse. Se consideran retrotransposones no autónomos, ya que dependen para propagarse de la retrotranscripción de su ARNm por una retrotranscriptasa presente en el medio.
LINE
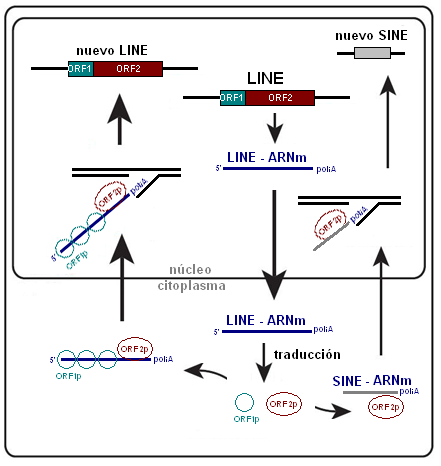
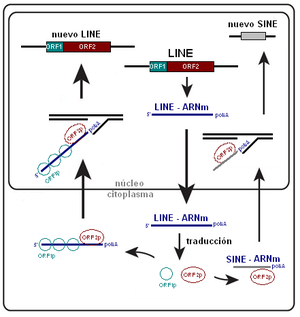 Esquema simplificado del mecanismo de retrotransposición de un elemento LINE y un SINE. Un elemento LINE es transcrito produciendo un ARNm que sale del núcleo celular. En el citoplasma se traduce en sus dos marcos de lectura abiertos generando ambas proteínas (véase el texto), que para simplificar se han representado como ORF1p y ORF2p. Ambas permiten retrotranscribir el ARNm del LINE y de otros retrotransposones no autónomos, como SINEs y pseudogenes procesados. Durante la retrotranscripción la nueva secuencia de ADN se integra en otro punto del genoma.
Esquema simplificado del mecanismo de retrotransposición de un elemento LINE y un SINE. Un elemento LINE es transcrito produciendo un ARNm que sale del núcleo celular. En el citoplasma se traduce en sus dos marcos de lectura abiertos generando ambas proteínas (véase el texto), que para simplificar se han representado como ORF1p y ORF2p. Ambas permiten retrotranscribir el ARNm del LINE y de otros retrotransposones no autónomos, como SINEs y pseudogenes procesados. Durante la retrotranscripción la nueva secuencia de ADN se integra en otro punto del genoma.Acrónimo del inglés Long Interspersed Nuclear Elements (Elementos nucleares dispersos largos). Constituyen el 20% del genoma humano. La familia de mayor importancia cuantitativa es LINE-1 o L1 que es una secuencia de 6 kb repetida unas 800.000 veces de modo disperso por todo el genoma, aunque la gran mayoría de las copias es incompleta al presentar el extremo 5' truncado por una retrotranscripción incompleta. Así, se estima que hay unas 5.000 copias completas de L1, sólo 90 de las cuales son activas,[10] estando el resto inhibidas por metilación de su promotor.
Su riqueza en G+C es del 42%,[10] próxima a la media del genoma (41%) y se localizan preferentemente en las bandas G de los cromosomas. Poseen además un promotor de la ARN polimerasa II.
Los elementos LINE completos son codificantes. En concreto LINE-1 codifica dos proteínas:
- Proteína de unión a ARN (’’RNA-binding protein’’): codificada por el marco de lectura abierto 1 (ORF1, acrónimo del inglés ‘’Open reading Frame 1’’)
- Enzima con actividad retrotranscriptasa y endonucleasa: codificada por el ORF2.
Por lo tanto, se consideran retrotransopsones autónomos, ya que codifican las proteínas que necesitan para propagarse. La ARN polimerasa II presente en el medio transcribe el LINE, y este ARNm se traduce en ambos marcos de lectura produciendo una retrotranscriptasa que actúa sobre el ARNm generando una copia de ADN del LINE, potencialmente capaz de insertarse en el genoma. Asimismo estas proteínas pueden ser utilizadas por pseudogenes procesados o elementos SINE para su propagación.
Diversos estudios han mostrado que las secuencias LINE pueden tener importancia en la regulación de la expresión génica, habiéndose comprobado que los genes próximos a LINE presentan un nivel de expresión inferior. Esto es especialmente relevante porque aproximadamente el 80% de los genes del genoma humano contiene algún elemento L1 en sus intrones.[10]
HERV
Acrónimo de Human endogenous retrovirus (retrovirus endógenos humanos). Los retrovirus son virus cuyo genoma está compuesto por ARN, capaces de retrotranscribirse e integrar su genoma en el de la célula infectada. Así, los HERV son copias parciales del genoma de retrovirus integrados en el genoma humano a lo largo de la evolución de los vertebrados, vestigios de antiguas infecciones retrovirales que afectaron a células de la línea germinal. Algunas estimaciones establecen que hay unas 98.000[12] secuencias HERV, mientras que otras afirman que son más de 400.000.[10] En cualquier caso, se acepta que en torno al 5-8% del genoma humano está constituido por genomas antiguamente virales. El tamaño de un genoma retroviral completo es de en torno a 6-11 kb, pero la mayoría de los HERV son copias incompletas.
A lo largo de la evolución estas secuencias sin interés para el genoma hospedador han ido acumulando mutaciones sin sentido y deleciones que los han inactivado. Aunque la mayoría de las HERV tienen millones de años de antigüedad, al menos una familia de retrovirus se integró durante la divergencia evolutiva de humanos y chimpancés, la familia HERV-K(HML2), que supone en torno al 1% de los HERV.
Transposones de ADN
Bajo la denominación de transposones a veces se incluyen los retrotransposones, tales como los pseudogenes procesados, los SINEs y los LINEs. En tal caso se habla de transposones de clase I para hacer referencia a los retrotransposones, y de clase II para referirse a transposones de ADN, a los que se dedica el presente apartado.
Los transposones de ADN completos poseen la potencialidad de autopropagarse sin un intermediario de ARNm seguido de retrotranscripción. Un transposón contiene en gen de una enzima transposasa, flanqueado por repeticiones invertidas. Su mecanismo de transposición se basa en cortar y pegar, moviendo su secuencia a otra localización distinta del genoma. Los distintos tipos de transposasas actúan de modo diferente, habiendo algunas capaces de unirse a cualquier parte del genoma mientras que otras se unen a secuencias diana específicas. La transposasa codificada por el propio transposón lo extrae realizando dos cortes flanqueantes en la hebra de ADN, generando extremos cohesivos, y lo inserta en la secuencia diana en otro punto del genoma. Una ADN polimerasa rellena los huecos generados por los extremos cohesivos y una ADN ligasa restablece los enlaces fosfodiéster, recuperando la continuidad de la secuencia de ADN. Esto conlleva una duplicación de la secuencia diana en torno al transposón, en su nueva localización.
Se estima que el genoma humano contiene unas 300.000 copias[10] de elementos repetidos dispersos originados por transposones de ADN, constituyendo un 3% del genoma. Hay múltiples familias, de las que cabe destacar por su importancia patogénica por la generación de reordenaciones cromosómicas los elementos mariner, así como las familias MER1 y MER2.
Variabilidad
Si bien dos seres humanos del mismo sexo comparten un porcentaje elevadísimo (en torno al 99,9%)[10] de su secuencia de ADN, lo que nos permite trabajar con una única secuencia de referencia, pequeñas variaciones genómicas fundamentan buena parte de la variabilidad fenotípica interindividual. Una variación en el genoma, por sustitución, deleción o inserción, se denomina polimorfismo o alelo genético. No todo polimorfismo genético provoca una alteración en la secuencia de una proteína o de su nivel de expresión, es decir, muchos son silenciosos y carecen de expresión fenotípica.
SNPs
La principal fuente de variabilidad en los genomas de dos seres humanos procede de las variaciones en un sólo nucleótido, conocidas como SNPs (Single nucleotide polimorphisms), en las cuales se han centrado la mayor parte de los estudios. Dada su importancia, en la actualidad existe un proyecto internacional (International HapMap Project) para catalogar a gran escala los SNPs del genoma humano. En este contexto, la denominación de SNP frecuentemente se restringe a aquellos polimorfismos de un sólo nucleótido en los que el alelo menos frecuente aparece en al menos el 1% de la población.
Los SNP son marcadores tetralélicos, dado que en teoría en una posición puede haber cuatro nucleótidos distintos, cada uno de los cuales identificaría un alelo; sin embargo, en la práctica suelen presentar sólo dos alelos en la población. Se estima que la frecuencia de SNPs en el genoma humano es de un SNP cada 500-100 pares de bases,[10] de los que una parte relevante son polimorfismos codificantes, que causan la sustitución de un aminoácido por otro en una proteína.
Gracias a su abundancia y a que presentan una distribución aproximadamente uniforme en el genoma, han tenido gran utilidad como marcadores para los mapas de ligamiento, herramienta fundamental del Proyecto Genoma Humano. Además son fácilmente detectables a gran escala mediante el empleo de chips de ADN (comúnmente conocidos como microarrays).
Variación estructural
Este tipo de variaciones se refiere a duplicaciones, inversiones, inserciones o variantes en el número de copias de segmentos grandes del genoma (por lo general de 1000 nucléotidos o más). Estas variantes implican a una gran proporción del genoma, por lo que se piensa que son, al menos, tan importantes como los SNPs.[13]
A pesar de que este campo de estudio es relativamente nuevo (los primeros estudios a gran escala se publicaron en los años 2004 y 2005), ha tenido un gran auge, hasta el punto de que se ha creado un nuevo proyecto para estudiar este tipo de variantes en los mismos individuos en los que se basó el Proyecto HapMap.
Aunque aún quedan dudas acerca de las causas de este tipo de variantes, cada vez existe más evidencia a favor de que es un fenómeno recurrente que todavía continua moldeando y creando nuevas variantes del genoma.
Este tipo de variaciones han potenciado la idea de que el genoma humano no es una entidad estática, sino que se encuentra en constante cambio y evolución.
Enfermedades genéticas
La alteración de la secuencia de ADN que constituye el genoma humano puede causar la expresión anormal de uno o más genes, originando un fenotipo patológico. Las enfermedades genéticas pueden estar causadas por mutación de la secuencia de ADN, con afectación de la secuencia codificante (produciendo proteínas incorrectas) o de secuencias reguladoras (alterando el nivel de expresión de un gen), o por alteraciones cromosómicas, numéricas o estructurales. La alteración del genoma de las células germinales de un individuo se transmite frecuentemente a su descendencia. Actualmente el número de enfermedades genéticas conocidas es aproximadamente de 4.000, siendo la más común la fibrosis quística.
El estudio de las enfermedades genéticas frecuentemente se ha englobado dentro de la genética de poblaciones. Los resultados del Proyecto Genoma Humano son de gran importancia para la identificación de nuevas enfermedades genéticas y para el desarrollo de nuevos y mejores sistemas de diagnóstico genético, así como para la investigación en nuevos tratamientos, incluida la terapia génica.
Mutaciones
Las mutaciones génicas pueden ser:
- Sustituciones (cambios de un nucleótido por otro): Las sustituciones se denominan transiciones si suponen un cambio entre bases del mismo tipo químico, o transversiones si son un cambio purina (A, G)→pirimidina (C, T) o pirimidina→purina.
- Deleciones o inserciones: son respectivamente la eliminación o adición de una determinada secuencia de nucleótidos, de longitud variable. Las grandes deleciones pueden afectar incluso a varios genes, hasta el punto de ser apreciables a nivel cromosómico con técnicas de citogenética. Inserciones o deleciones de unas pocas pares de bases en una secuencia codificante pueden provocar desplazamiento del marco de lectura (frameshift), de modo que la secuencia de nucleótidos del ARNm se lee de manera incorrecta.
Las mutaciones génica pueden afectar a:
- ADN codificante: Si el cambio en un nucleótido provoca en cambio de un aminoácido de la proteína la mutación se denomina no sinónima. En caso contrario se denominan sinónimas o silenciosas (posible porque el código genético es degenerado). Las mutaciones no sinónimas asimismo se clasifican en mutaciones con cambio de sentido (missense) si provocan el cambio de un aminoácido por otro, mutaciones sin sentido (non-sense) si cambian un codón codificante por un codón de parada (TAA, TAG, TGA) o con ganancia de sentido si sucede a la inversa.
- ADN no codificante: Pueden afectar a secuencias reguladoras, promotoras o implicadas en el ayuste (splicing). Estas últimas pueden causar un erróneo procesamiento del ARNm, con consecuencias diversas en la expresión de la proteína codificada por ese gen.
Trastornos de un sólo gen
Son enfermedades genéticas causadas por mutación en un sólo gen, que presentan una herencia de tipo mendeliano, fácilmente predecible. En la tabla se resumen los principales patrones de herencia que pueden mostrar, sus características y algunos ejemplos.
Patrón hereditario Descripción Ejemplos Autosómico dominante Enfermedades que se manifiestan en individuos heterocigóticos. Es suficiente con una mutación en una de las dos copias (recuérdese que cada individuo posee un par de cada cromosoma) de un gen para que se manifieste la enfermedad. Los individuos enfermos generalmente tienen uno de sus dos progenitores enfermos. La probabilidad de tener descendencia afectada es del 50% dado que cada progenitor aporta uno de los cromosomas de cada par. Frecuentemente corresponden a mutaciones con ganancia de función (de modo que el alelo mutado no es inactivo sino que posee una nueva función que provoca el desarrollo de la enfermedad) o por pérdida de función del alelo mutado con efecto de dosis génica también conocido como haploinsuficiencia. Frecuentemente son enfermedades con baja penetrancia, es decir, sólo una parte de los individuos que portan la mutación desarrollan la enfermedad. Enfermedad de Huntington, Neurofibromatosis 1, Síndrome de Marfan, Cáncer colorrectal hereditario no polipósico Autosómico recesivo La enfermedad sólo se manifiesta en individuos homocigóticos recesivos, es decir, aquellos en los que ambas copias de un gen están mutadas. Son mutaciones que causan pérdida de función, de modo que la causa de la enfermedad es la ausencia de la acción de un gen. La mutación sólo en una de las dos copias es compensada por la existencia de la otra (cuando una sola copia no es suficiente se origina haploinsuficiencia, con herencia autosómica dominante). Habitualmente un individuo enfermo tiene ambos progenitores sanos pero portadores de la mutación (genotipo heterocigótico: Aa). En tal caso un 25% de la descendencia estará afectada. Fibrosis quística, Anemia falciforme, Enfermedad de Tay-Sachs, Atrofia muscular espinal Dominante ligado al X Las enfermedades dominantes ligadas al cromosoma X están causadas por mutaciones en dicho cromosoma, y presentan un patrón hereditario especial. Sólo unas pocas enfermedades hereditarias presentan este patrón. Las mujeres tienen mayor prevalencia de la enfermedad que los hombres, dado que reciben un cromosoma X de su madre y otro de su padre, cualquiera de los cuales puede portar la mutación. Los varones en cambio siempre reciben el cromosoma Y de su padre. Así, un varón enfermo (xY) tendrá todos sus hijos varones sanos (XY) y todas las hijas enfermas (Xx), mientras que una mujer enferma (Xx) tendrá un 50% de su descendencia enferma, independientemente del sexo. Algunas de estas enfermedades son letales en varones (xY), de modo que sólo existen mujeres enfermas (y varones con Síndrome de Klinefelter, XxY). Hipofosfatemia, Síndrome de Aicardi Recesivo ligado al X Las enfermedades recesivas ligadas al X también están causadas por mutaciones en el cromosoma X. Los varones están más frecuentemente afectados. Un varón portador siempre será enfermo (xY) dado que sólo posee un cromosoma X, que está mutado. Su descendencia serán varones sanos (XY) e hijas portadoras (Xx). Una mujer portadora, tendrá una descendencia compuesta por un 50% de hijas portadoras y un 50% de varones enfermos. Hemofilia A, Distrofia Muscular de Duchenne, Daltonismo, Distrofia muscular Alopecia androgénica Ligado a Y Son enfermedades causadas por mutación en el cromosoma Y. En consecuencia, sólo puede manifestarse en varones, cuya descendencia será del 100% de hijas sanas y el 100% de hijos varones enfermos. Dadas las funciones del cromosoma Y, frecuentemente estas enfermedades sólo causan infertilidad, que a menudo puede ser superada terapéuticamente. Infertilidad masculina hereditaria Mitocondrial Enfermedades causadas por mutación en genes del genoma mitocondrial. Dadas la particularidades de dicho genoma, su transmisión es matrilineal (el genoma mitocondrial se transfiere de madres a hijos). La gravedad de una mutación depende del porcentaje de genomas afectados en la población de mitocondrias, fenómeno denominado heteroplasmia (en contraste con heterocigosis), que varía por segregación mitótica asimétrica. Neuropatía óptica hereditaria de Leber (LHON) Trastornos poligénicos y multifactoriales
Otras alteraciones genéticas pueden ser mucho más complejas en su asociación con un fenotipo patológico. Son las enfermedades multifactoriales o poligénicas, es decir, aquellas que están causadas por la combinación de múltiples alelos genotípicos y de factores exógenos, tales como el ambiente o el estilo de vida. En consecuencia no presentan un patrón hereditario claro, y la diversidad de factores etiológicos y de riesgo dificulta la estimación del riesgo, el diagnóstico y el tratamiento.
Algunos ejemplos de enfermedades multifactoriales con etiología parcialmente genética son:
Alteraciones cromosómicas
Las alteraciones genéticas pueden producirse también a escala cromosómica (cromosomopatías), causando severos trastornos que afectan a múltiples genes y que en muchas ocasiones son letales provocando abortos prematuros. Frecuentemente están provocadas por un error durante la división celular, que sin embargo no impide su conclusión. Las alteraciones cromosómicas reflejan una anormalidad en el número o en la estructura de los cromosomas, por lo que se clasifican en numéricas y estructurales. Provocan fenotipos muy diversos, pero frecuentemente presentan unos rasgos comunes:
- Retraso mental y retraso del desarrollo.
- Alteraciones faciales y anomalías en cabeza y cuello.
- Malformaciones congénitas, con afectación preferente de extremidades, corazón, etc.
Numéricas
Frecuencias de aneuploidías por cada 1000 nacidos vivos.[10] Aneuploidía Frecuencia
(/1000)Síndrome Trisomía 21 1,5 de Down Trisomía 18 0,12 de Edwards Trisomía 13 0,07 de Patau Monosomía X 0,4 de Turner XXY 1,5 de Klinefelter XYY 1,5 del XYY Es una alteración del número normal de cromosomas de un individuo, que normalmente presenta 23 pares de cromosomas (46 en total), siendo cada dotación cromosómica de un progenitor (diploidía). Si la alteración afecta a un sólo par de cromosomas se habla de aneuploidía, de manera que puede haber un sólo cromosoma (monosomía) o más de dos (trisomía, tetrasomía...). Un ejemplo de gran prevalencia es la trisomía 21, responsable del Síndrome de Down. Si por el contrario la alteración afecta a todos los cromosomas se habla de euploidías, de manera que en teoría el individuo tiene una sola dotación cromosómica (haploidía, 23 cromosomas en total) o más de dos dotaciones (triploidía: 69 cromosomas; tetraploidía: 92 cromosomas...). En la práctica las euploidías causan letalidad embronaria (abortos) siendo muy pocos los nacidos vivos, y fallecen muy tempranamente. Las aneuploidías son mayoritariamente letales, salvo las trisomías de los cromosomas 13, 18, 21, X e Y (XXY, XYY), y la monosomía del cromosoma X. En la tabla se muestran las frecuencias de nacidos vivos con estas alteraciones.
Estructurales
Se denominan así las alteraciones en la estructura de los cromosomas, tales como las grandes deleciones o inserciones, reordenaciones del material genético entre cromosomas... detectables mediante técnicas de citogenética.
- Deleciones: eliminación de una porción del genoma. Algunos trastornos conocidos son el Síndrome de Wolf-Hirschhorn por deleción parcial del brazo corto del cromosoma 4 (4p), y el Síndrome de Jacobsen o deleción 11q terminal.
- Duplicaciones: una región considerable de un cromosoma se duplica. Un ejemplo es la enfermedad de Charcot-Marie-Tooth tipo 1A, que puede ser causada por duplicación del gen codificante de la proteína mielínica periférica 22 (PMP22) en el cromosoma 17.
- Translocaciones: cuando una porción de un cromosoma se transfiere a otro cromosoma. Hay dos tipos principales de translocaciones: la translocación recíproca, en la que se intercambian segmentos de dos cromosomas distintos, y la translocación Robertsoniana, en la que dos cromosomas acrocéntricos (13, 14, 15, 21, 22) se fusionan por sus centrómeros (fusión céntrica).
- Inversiones: una parte del genoma se rompe y se reorienta en dirección opuesta antes de reasociarse, con lo que dicha secuencia aparece invertida. Pueden ser paracéntricas (si afectan sólo a una brazo) o pericéntricas (si la secuencia invertida incluye el centrómero).
- Cromosomas en anillos: una porción del genoma se rompe y forma un anillo por circularización. Esto puede ocurrir con pérdida de material o sin pérdida de material.
- Isocromosomas: cromosomas simétricos, con sus dos brazo idénticos por deleción de uno de los brazos y duplicación del otro. El más habitual es el isocromosoma X, en el que se pierde el brazo corto del cromosoma X, originando fenotipos de Síndrome de Turner.
Los síndromes de inestabilidad cromosómica son un grupo de trastornos caracterizados por una gran inestabilidad de los cromosomas, que sufren con gran frecuencia alteraciones estructurales. Están asociados con un aumento de la malignidad de neoplasias.
Evolución
Los estudios de genómica comparada se basan en comparación de secuencias genómicas a gran escala, generalmente mediante herramientas bioinformáticas. Dichos estudios permiten ahondar en el conocimiento de aspectos evolutivos de escala temporal y espacial muy diversa, desde el estudio de la evolución de los primeros seres vivos hace miles de millones de años o las radiaciones filogenéticas en mamíferos, hasta el estudio de las migraciones de seres humanos en los últimos 100.000 años, que explican la actual distribución de las distintas razas humanas.
Genómica comparada entre distintas especies
Los estudios de genómica comparada con genomas de mamíferos sugieren que aproximadamente el 5% del genoma humano se ha conservado evolutivamente en los últimos 200 millones de años; lo cual incluye la gran mayoría de los genes y secuencias reguladoras. Sin embargo, los genes y las secuencias reguladoras actualmente conocidas suponen sólo el 2% del genoma, lo que sugiere que la mayor parte de la secuencia genómica con gran importancia funcional es desconocida. Un porcentaje importante de los genes humanos presenta un alto grado de conservación evolutiva. La similitud entre el genoma humano y el del chimpancé (Pan troglodytes) es del 98,77%. En promedio, una proteína humana se diferencia de su ortóloga de chimpancé en tan sólo dos aminoácidos, y casi un tercio de los genes tiene la misma secuencia. Una diferencia importante entre los dos genomas es el cromosoma 2 humano, que es el producto de una fusión entre los cromosomas 12 y 13 del chimpancé[14]
Otra conclusión de la comparación del genoma de distintos primates es la notable pérdida de genes de receptores olfativos que se ha producido paralelamente al desarrollo de la visión en color (tricrómica) durante la evolución de primates.[15]
Genómica comparada entre genomas humanos
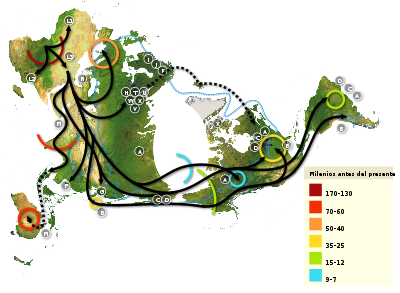 Mapa de las migraciones humanas creado a partir de genómica comparada con los genomas mitocondriales de individuos actuales. Los números de la leyenda representan miles de años antes del presente. La línea azul rayada delimita el área cubierta de hielo o de tundra durante la última glaciación. Las letras englobadas por círculos indican los halogrupos de ADN mitocondrial; los halogrupos se usan para definir subpoblaciones genéticas, que frecuentemente tienen una correlación geográfica. Los principales halogrupos de ADNmt son: Africa: L, L1, L2, L3. Oriente próximo: J, N. Europa meridional: J, K. Europa (general): H, V. Europa septentrional: T, U, X. Asia: A, B, C, D, E, F, G (en el dibujo: M está compuesta por C, D, E, y G). Nativos Americanos: A, B, C, D y a menudo X. Véase el artículo: Haplogrupos de ADN mitocondrial humano.
Mapa de las migraciones humanas creado a partir de genómica comparada con los genomas mitocondriales de individuos actuales. Los números de la leyenda representan miles de años antes del presente. La línea azul rayada delimita el área cubierta de hielo o de tundra durante la última glaciación. Las letras englobadas por círculos indican los halogrupos de ADN mitocondrial; los halogrupos se usan para definir subpoblaciones genéticas, que frecuentemente tienen una correlación geográfica. Los principales halogrupos de ADNmt son: Africa: L, L1, L2, L3. Oriente próximo: J, N. Europa meridional: J, K. Europa (general): H, V. Europa septentrional: T, U, X. Asia: A, B, C, D, E, F, G (en el dibujo: M está compuesta por C, D, E, y G). Nativos Americanos: A, B, C, D y a menudo X. Véase el artículo: Haplogrupos de ADN mitocondrial humano.Durante décadas las únicas evidencias que permitían profundizar en el conocimiento del origen y la expansión del Homo sapiens han sido los escasos hallazgos arqueológicos. Sin embargo, en la actualidad, los estudios de genómica comparada a partir de genomas de individuos actuales de todo el mundo, están aportando información muy relevante. Su fundamento básico consiste en identificar un polimorfismo, una mutación, que se asume que se originó en un individuo de una población ancestral, y que ha heredado toda su descendencia hasta la actualidad. Además, dado que las mutaciones parecen producirse a un ritmo constante, puede estimarse la antigüedad de una determinada mutación en base al tamaño del haplotipo en el que se sitúa, es decir, el tamaño de la secuencia conservada que flanquea la mutación. Esta metodología se ve complicada por el fenómeno de recombinación entre los pares de cromosomas de un individuo, procedentes de sus dos progenitores. Sin embargo, hay dos regiones en las que no existe dicho inconveniente porque presentan una herencia uniparental: el genoma mitocondrial (de herencia matrilineal), y el cromosoma Y (de herencia patrilineal).
En las últimas décadas, los estudios de genómica comparada basada en el genoma mitocondrial, y en menor medida en el cromosoma Y, han reportado conclusiones de gran interés. En diversos estudios se ha trazado la filogenia de estas secuencias, estimándose que todos los seres humanos actuales comparten un antepasado femenino común que vivió en África hace unos 150.000 años. Por su parte, por razones aún poco conocidas, la mayor convergencia del ADN del cromosoma Y establece que el antepasado masculino común más reciente data de hace unos 60.000 años. Estos individuos han sido bautizados como Eva mitocondrial e Y-cromosoma Adan.
La mayor diversidad de marcadores genéticos y en consecuencia, los haplotipos de menor longitud, se han hallado en África. Todo el resto de la población mundial presenta sólo una pequeña parte de estos marcadores, de modo que la composición genómica del resto de la población humana actual es sólo un subconjunto de la que puede apreciarse en África. Esto induce a afirmar que un pequeño grupo de seres humanos (quizá en torno a un millar) emigró del continente africano hacia las costas de Asia occidental, hace unos 50.000-70.000 años, según estudios basados en el genoma mitocondrial. Hace unos 50.000 años alcanzaron Australia y hace en torno a 40.000-30.000 años otras subpoblaciones colonizaron Europa occidental y el centro de Asia. Asimismo, se estima que hace 20.000-15.000 años alcanzaron el continente americano a través del estrecho de Bering (el nivel del mar era menor durante la última glaciación, o glaciación de Würm o Wisconsin), poblando Sudamérica hace unos 15.000-12.000 años. No obstante, estos datos sólo son estimaciones, y la metodología presenta ciertas limitaciones. En la actualidad, la tendencia es combinar los estudios de genómica comparada basados en el ADN mitocondrial con análisis de la secuencia del cromosoma Y.
Genoma mitocondrial
Es el genoma propio de las mitocondrias de células eucariotas. La mitocondria es un orgánulo subcelular esencial en el metabolismo aerobio u oxidativo de las células eucariotas. Su origen es endosimbionte, es decir, antiguamente fueron organismos procariotas independientes captados por una célula eucariota ancestral, con la que desarrollaron una relación simbiótica. Las características de su genoma, por tanto, son muy semejantes a las de un organismo procariota actual, y su código genético es ligeramente distinto al considerado universal. Para adaptarse al nicho intracelular y aumentar su tasa de replicación, el genoma mitocondrial se ha ido reduciendo sustancialmente a lo largo de su coevolución, presentando en la actualidad un tamaño de 16.569 pares de bases. Así, la gran mayoría de las proteínas localizadas en las mitocondrias (~1500 en mamíferos) están codificadas por el genoma nuclear (al que hacen referencia todos los apartados anteriores), de modo que muchos de estos genes fueron transferidos de la mitocondria al núcleo celular durante la coevolución de la célula eucariota. En la mayoría de mamíferos, sólo la hembra transmite al zigoto sus mitocondrias, por lo que presentan, como ya se ha dicho, un patrón hereditario matrilineal. En general una célula humana media contiene 100-10.000 copias del genoma mitocondrial por cada célula, a razón de unas 2-10 moléculas de ADN por mitocondria.
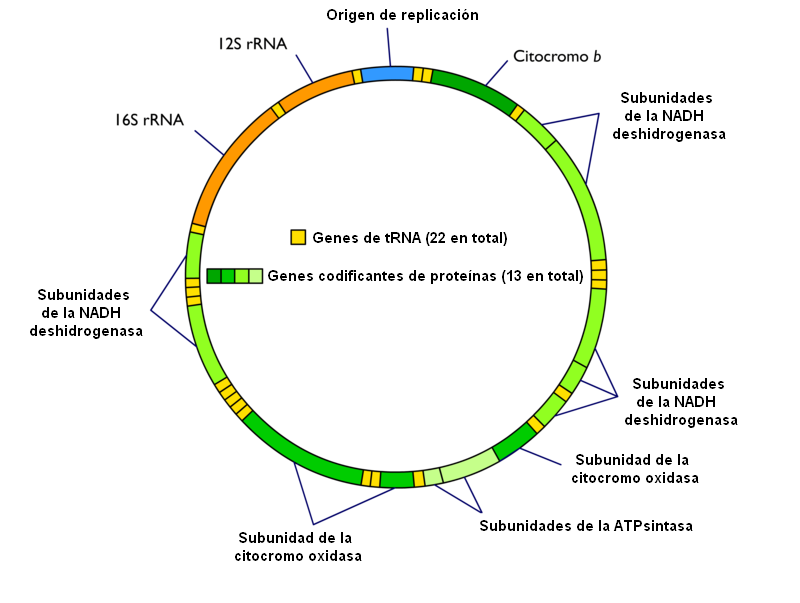
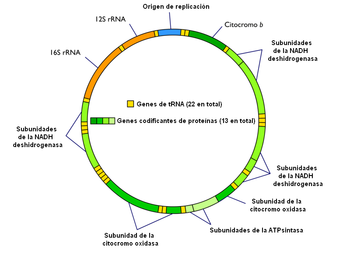 Diagrama simplificado del genoma mitocondrial. Pueden apreciarse los 37 genes y la secuencia origen de replicación no codificante. En este esquema no se señala la cadena ligera y la pesada.
Diagrama simplificado del genoma mitocondrial. Pueden apreciarse los 37 genes y la secuencia origen de replicación no codificante. En este esquema no se señala la cadena ligera y la pesada.El genoma mitocondrial posee 37 genes:[10]
- 13 genes codificantes de proteínas: codifican 13 polipéptidos que forman parte de los complejos multienzimáticos de la fosforilación oxidativa (sistema OXPHOS). Son 7 subunidades del Complejo I (NADH deshidrogenasa), una subunidad del complejo III (citocromo b), 3 subunidades del Complejo IV (citocromo oxidasa) y 2 subunidades del Complejo V (ATPsintasa).
- 2 genes ARNr, que codifican las dos subunidades del ARN ribosómico de la matriz mitocondrial.
- 22 genes ARNt, que codifican los 22 ARN transferentes necesarios para la síntesis proteica en la matriz mitocondrial.
Al contrario de lo que sucedía con el genoma nuclear, donde sólo el 1,5% era codificante, en el genoma mitocondrial el 97% corresponde a secuencias codificantes. Es una única molécula de ADN doble hebra circular. Una de las hemihebras recibe el nombre de cadena pesada o cadena H, y contiene 28 de los 37 genes (2 ARNr, 14 ARNt y 12 polipéptidos). La hemihebra complementaria (cadena ligera o L) codifica los 9 genes restantes. En ambas cadenas, los genes de los ARNt aparecen distribuidos entre dos genes ARNr o codificantes de proteínas, lo cual es de gran importancia para el procesamiento del ARN mitocondrial.
Véase también
- ADN | Gen | Transcripción | Traducción | Código genético | Genética
- Historia de la genética | Gregor Mendel | James D. Watson | Francis Crick | Jérôme Lejeune | Francis Collins
Referencias
- ↑ International Human Genome Sequencing Consortium (2004). «Finishing the euchromatic sequence of the human genome.». Nature 431 (7011): pp. 931-45. PMID 15496913. [1]
- ↑ International Human Genome Sequencing Consortium (2001). «Initial sequencing and analysis of the human genome.». Nature 409 (6822): pp. 860-921. PMID 11237011. [2]
- ↑ Watson, JD, Baker TA, Bell SP, Gann A, Levine M, Losick R. (2004). “Ch9-10”, Molecular Biology of the Gene, 5th ed., Peason Benjamin Cummings; CSHL Press.
- ↑ Sequenced percentages are based on fraction of euchromatin portion, as the Human Genome Project goals called for determination of only the euchromatic portion of the genome. Telomeres, centromeres, and other heterochromatic regions have been left undetermined, as have a small number of unclonable gaps. See http://www.ncbi.nlm.nih.gov/genome/seq/ for more information on the Human Genome Project.
- ↑ The ENCODE Project Consortium. (2007). «Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project.». Nature 447 (7146): pp. 799-816. PMID: 17571346..
- ↑ Mark B. Gerstein, Can Bruce, Joel S. Rozowsky, Deyou Zheng, Jiang Du, Jan O. Korbel, Olof Emanuelsson, Zhengdong D. Zhang, Sherman Weissman, and Michael Snyder (2007). «What is a gene, post-ENCODE? History and updated definition». Genome Research 17: pp. 669-681. [3] Versión completa accesible gratuitamente.
- ↑ a b http://www.unav.es/genetica/GH/cap5.html Página web con recientes actualizaciones del libro del Dr. Novo. Incluye un video en castellano sobre la redefinición del concepto de gen[4].
- ↑ Loots G, Locksley R, Blankespoor C, Wang Z, Miller W, Rubin E, Frazer K (2000). «Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons.». Science 288 (5463): pp. 136-40. PMID 10753117. Summary
- ↑ Nei M, Xu P, Glazko G (2001). «Estimation of divergence times from multiprotein sequences for a few mammalian species and several distantly related organisms.». Proc Natl Acad Sci U S A 98 (5): pp. 2497-502. PMID 11226267. http://www.pnas.org/cgi/content/full/051611498.
- ↑ a b c d e f g h i j k l m n ñ o Novo Villaverde, F.J. (2007). Genética Humana. Madrid: Pearson. ISBN 8483223598. (Recomendado)
- ↑ Torrents D., Suyama M., Zdobnov E. and Bork p. (2003). «A Genome-Wide Survey of Human Pseudogenes.». Genome Research 13 (12): pp. 2559-2567. PMID 14656963. [5]
- ↑ Robert Belshaw, (2004). "Long-term reinfection of the human genome by endogenous retroviruses" Proc Natl Acad Sci U S A. 2004 April 6; 101(14): 4894–4899
- ↑ Feuk L., Carson A. R. y Scherer S. W. (2006). «Structural Variation in the human Genome.». Nature Reviews Genetics 7 (2): pp. 85-97. PMID 16418744.[6]
- ↑ "Human chromosome 2 resulted from a fusion of two ancestral chromosomes that remained separate in the chimpanzee lineage" The Chimpanzee Sequencing and Analysis Consortium (2005). «Initial sequence of the chimpanzee genome and comparison with the human genome.». Nature 437 (7055): pp. 69-87. PMID 16136131.
"Large-scale sequencing of the chimpanzee genome is now imminent."Olson M, Varki A (2003). «Sequencing the chimpanzee genome: insights into human evolution and disease.». Nat Rev Genet 4 (1): pp. 20-8. PMID 12509750. - ↑ "Our findings suggest that the deterioration of the olfactory repertoire occurred concomitant with the acquisition of full trichromatic color vision in primates." Gilad Y, Wiebe V, Przeworski M, Lancet D, Pääbo S (2004). «Loss of olfactory receptor genes coincides with the acquisition of full trichromatic vision in primates.». PLoS Biol 2 (1): pp. E5. PMID 14737185.
Enlaces externos
En español
- Asociaciones, sociedades e institutos:
- Asociación Española de Genética Humana
- Instituto de Genética Humana (Universidad Javeriana, Fac. Medicina)
Artículos
- Recursos educativos y artículos divulgativos:
- El genoma humano (UNNE)
- Genética humana (Junta de Andalucía)
- Selección de enlaces en castellano
- La investigación sobre el genoma humano
- El significado biológico del mensaje genético escrito en el genoma
- INMEGEN: Mapa genético completo de algunos pueblos nativos mexicanos
En inglés
- Secuencia del genoma humano. Science 2001;291;5507;1304-51.Texto completo
- The National Human Genome Research Institute
- National Library of Medicine human genome viewer . (Recomendado). Interfaz de acceso a la secuencia del genoma humano.
- UCSC Genome Browser. (Recomendado). Interfaz de acceso a la secuencia del genoma humano.
- OMIM: Online Mendelian Inheritance in Man. (Recomendado). Base de datos de enfermedades genéticas.
- Human Genome Project.
- Sabancı University School of Languages Podcasts What makes us different from chimpanzees? by Andrew Berry (MP3 file)
- The National Office of Public Health Genomics
- The Genographic Project de National Geographic
Categorías:- Genomas secuenciados
- Genética


Wikimedia foundation. 2010.