- Enfermedad de Alexander
-
Enfermedad de Alexander 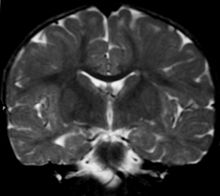
Imagen de una leucodistrofia en una niña de 18 meses. Posible Enfermedad de AlexanderClasificación y recursos externos CIE-10 G37.8 CIE-9 331.89 OMIM 2034450 Sinónimos -ALX
-AxD
-Leucodistrofia desmielinogénica
-Degeneración astrocítica fibrinoide
-Leucodistrofia con fibras de Rosenthal
-Panneuropatía hialina Aviso médico
Aviso médico La enfermedad de Alexander es una enfermedad genética extremadamente rara, normalmente de aparición en la infancia y perteneciente al grupo de las leucodistrofias. Este grupo de enfermedades neurológicas se caracteriza por la destrucción progresiva de la sustancia blanca del cerebro. La enfermedad de Alexander se manifiesta por la aparición de retraso mental y anormalidades físicas, en especial macrocefalia, por la presencia de fibras de Rosenthal y patrones de neuroimagen característicos. La enfermedad progresa hasta un desenlace mortal en la mayor parte de los casos. La enfermedad recibe el nombre por el patólogo neozelandés William Stuart Alexander, quien describió el síndrome en 1949 junto con la profesora Dorothy Rusell en el London Hospital.[1]
Contenido
Epidemiología
La prevalencia de la enfermedad es desconocida. La mayor parte de los casos se dan de forma esporádica, sin que exista un historial familiar de propensión a la enfermedad. Entre otras cosas, esto significa que los padres que tienen un hijo con la enfermedad, tienen una probabilidad muy baja de que siguientes hijos la tengan, sin embargo, existen algunas familias con más de un hijo afectado. Es posible que exista una heredabilidad de la propensión a padecer mutaciones "de novo" en general en la descendencia. Así mismo, existen dos casos de familias en que la enfermedad se hereda de forma dominante.[2] En ambos, la aparición es posterior a los 25 años. Hasta 2005 existían tan solo 57 casos descritos. Se han publicado 500 casos desde su descripción en 1949.[3] La enfermedad es más frecuente en mujeres (proporción 3:1).es una enfermedad degenerativa
Etiología
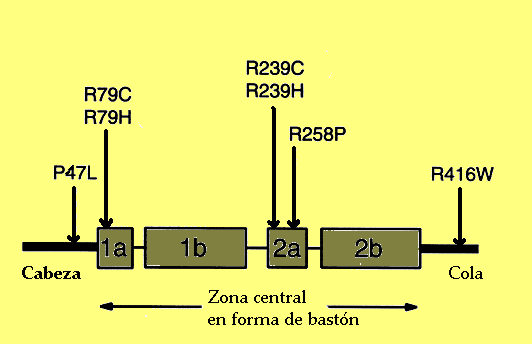
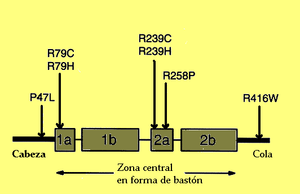 Localización de las principales mutaciones en la PAFG. De ellas, la P47L podría no estar asociada a la enfermedad. Los rectángulos representan los dominios α-hélice de la proteína.
Localización de las principales mutaciones en la PAFG. De ellas, la P47L podría no estar asociada a la enfermedad. Los rectángulos representan los dominios α-hélice de la proteína.
En principio, se observó la acumulación en el cerebro en torno a los astrocitos y a la barrera hematoencefálica de unas estructuras características eosinófilas y vermiformes que se llamaron fibras de Rosenthal, en honor al patólogo alemán Werner Rosenthal quien las describió en 1989 en la siringomielia. Estas estructuras eran ricas en PAFG y en una proteína de shock término llamada α-B-Cristalina.
La alteración subyacente fue caracterizada en 2001 por Messing y colaboradores.[4] Se trata de una enfermedad de carácter genético autosómica dominante, originada en la mayor parte de los casos por una mutación puntual de novo en el gen de la proteína ácida fibrilar glial. Este gen está situado en la banda q21 del cromosoma 17 (17 q21). Li y colaboradores observaron que en 24 de 28 casos de su estudio, el cromosoma portador es el paterno, con lo que parece que la mutación se da mucho más en la espermiogénesis que durante el desarrollo embrionario. Parece ser que no hay una relación con la edad del padre.[5] Se han descrito más de 49 mutaciones o alelos patológicos que pueden desencadenar la enfermedad:- Diana Rodríguez y colaboradores encontraron en nueve pacientes mutaciones en el dominio central en forma de bastón de la proteína, habitualmente en los segmentos 1A, 2A y 2B. La mayor parte daban cambios en residuos arginina (cuatro en la posición R79H, cuatro en R239C y una en R239H, 2R88C y 1R88S, estas dos últimas desconocidas hasta ese momento). Los dos casos restantes del estudio afectaban a otros aminoácidos (1L76F y 1N77Y). El aminoácido afectado parecía determinar la severidad del curso de la enfermedad.[6]
- En 2007 Meins y colaboradores detectaron otras mutaciones puntuales en otro dominio (cuya posición está próxima al C terminal de la proteína) que determinaban la aparición temprana de la enfermedad, en concreto A364 V y Y366C. Se observo que mutaciones idénticas en otros filamentos intermedios, por ejemplo en la queratina, daban lugar a problemas similares, con lo que se sugiere que estas secuencias son críticas para la estabilidad de estos filamentos.[7]
Patogenia
Hasta la fecha, el mecanismo propuesto más aceptado para explicar la enfermedad sería el siguiente:[8]
- La acumulación de proteína ácida fibrilar glial (PAFG) y la consiguiente formación de agregados característicos, denominados fibras de Rosenthal en varios tipos celulares, y en especial los astrocitos. Parece que la acumulación se debe a una ganancia de función por causa de la mutación que bloquea parcialmente el ensamblaje de los filamentos de PAFG.[9]
- Secuestro posterior de ubiquitina y las proteínas chaperonas α-B-cristalina y HSP27 en las fibras de Rosenthal.
- Activación tanto de la proteína Jnk como de la respuesta de estrés.
Anatomía patológica
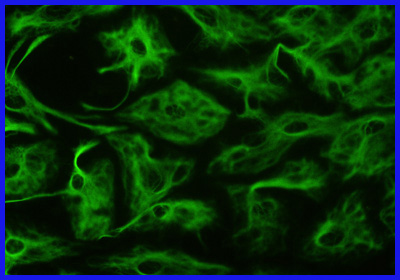
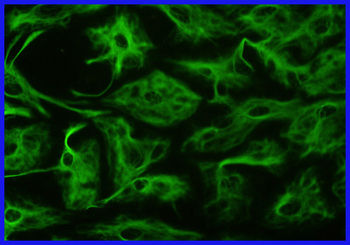 Astrocitos fibrosos visibles mediante técnicas de inmunohistoquímica gracias a anticuerpos quiméricos (unidos a proteína verde fluorescente) contra la proteína ácida fibrilar glial.
Astrocitos fibrosos visibles mediante técnicas de inmunohistoquímica gracias a anticuerpos quiméricos (unidos a proteína verde fluorescente) contra la proteína ácida fibrilar glial.La enfermedad de Alexander es primariamente una alteración de los astrocitos, que forman parte de las células de soporte de las neuronas (neuroglía). Entre los astrocitos, los más comunes en la sustancia blanca son los de tipo fibroso.[10] Estos últimos se caracterizan porque en su citoesqueleto contienen un filamento intermedio, la ya mencionada PAFG, que al mutar construye una estructura proteica defectuosa. Esta se almacena junto con la ubiquitina y otras dos proteínas de shock térmico, las ya conocidas fibras de Rosenthal. Se pueden localizar en todo el sistema central, tanto en el cerebro como en la médula espinal, pero en especial en la vecindad de los vasos sanguíneos de la superficie del cerebro. Las imágenes por microscopía electrónica muestran un vínculo estrecho entre las fibras de Rosenthal y los filamentos intermedios. Además existe desmielinización histológicamente hablando en los afectados tardíos, o ausencia de mielinización en los niños. Se encuentran afectadas igualmente las fibras sensoriales y las motoras. En la aparición temprana con macrocefalia es común la degeneración de la sustancia blanca y a veces se acompaña también de hidrocefalia.
Sin embargo, las áreas desmielinizadas no coinciden con la distribución de las fibras de Rosenthal, por ello parece que la desmielinización y la aparición de fibras parecen ser manifestaciones independientes de la enfermedad. No obstante se asume que la desmielinización se produce por la degeneración final de los astrocitos.
Tabla1: CUADRO CLÍNICO de enfermos diagnosticados mediante MRI de enfermedad de Alexander en una muestra de 5 y 14 pacientes respectivamente, según van der Knaap y otros[11] Signos observados Variante infantil variante juvenil Nº de pacientes 5 14 Edad de primeras manifestaciones nacimiento a 4 semanas <2 años Retraso desarrollo motor severo(5) severo(2)moderado(11)no observado(1) Principal indicador motor ninguno(3)asir(2) marcha(12)sentarse(2) Retraso desarrollo mental Severo(5) moderado(13) Problemas de conducta 2 9 Epilepsia Severa (5) Moderada (11) Probemas de alimentación 5 12 Vómitos 5 6 Dificultades para tragar/asfixia 4 11 ganancia peso insuf 5 5 Problemas de habla 2, no aplicable en 3 14 Macrocefalia 4,98º percentil en 1 9,98º percentil en 2/no obs. en 3 Pobre contacto visual 5 0 Hipotonía Generalizada (2)
axial(3)Generalizada (3) Hipertonía de miembros 3 8 Hiperreflexia 5 14 Ataxiacerebelar No comprobable 12 Signos extrapiramidales 3 3 Deterioro rápido(3)moderado(2) lento(11)moderado(1) Edad de fallecimiento 4 meses a 2¹/2 años 7-18 años en 4 casos Cuadro clínico
La enfermedad tiene varias formas que difieren en su cuadro clínico y en la edad de aparición, teniendo todas ellas el rasgo distintivo de la degeneración fibrinoide de los astrocitos con fibras de Rosenthal. Desde 1976 se reconocen tres formas de la enfermedad:[12]
- Forma infantil:
-
-
- Aparece desde el nacimiento hasta los 2 años.
- Es la más común.
- Puede cursar con o sin macroencefalia, aunque lo más corriente es que aparezca (Rodríguez, 2001).
- Se observan ataques y retraso o involución en el desarrollo. La función motora se deteriora progresivamente hasta la cuadriparesis y espasticidad.
- Hidrocefalia, en ocasiones asociada a estenosis del acueducto de Silvio.[13] Aunque no se ha observado asociación entre la estenosis y la acumulación de fibras de Rosenthal.
- Retraso mental profundo en la mayoría de los casos, aunque a veces no se ha observado.
- Ataxia y ataques epilépticos.
- vómitos y elevada tensión intracraneal.
-
- Forma juvenil:[14]
-
-
- Comienza en la edad escolar, a los 9,5 años de media
- La sintomatología consiste principalmente en paraplejia espástica y signos bulbares progresivos.
- Normalmente se preservan las funciones cognitivas.
- Dificultades para tragar o hablar, vómitos, ataxia y/o espasticidad.
- Puede darse cifoscoliosis
- Mientras la forma infantil afecta generalmente al cerebro, la juvenil afecta más al tronco cerebral. Hay muchas fibras de Rosenthal, pero la desmielinización es menos importante que en la forma infantil.
-
- Forma adulta:
-
-
- Es la forma más benigna y rara de la enfermedad. Se han observado casos de aparición hasta los 36 años. Recuerda a la esclerosis múltiple o un tumor.
- La ataxia es muy frecuente, así como dificultades de habla, deglución y problemas en el sueño.
- Los síntomas son similares a la enfermedad juvenil, pero más suaves.
-
Diagnóstico
El diagnóstico había sido durante mucho tiempo difícil, debido a que la mayoría de los signos podían darse también en otras leucodistrofias, de modo que la prueba diagnóstica de confirmación consistía en una biopsia cerebral que revelara las fibras de Rosenthal. En ocasiones esto se efectuaba post mórtem en la necropsia. Sin embargo, esta prueba resulta bastante invasiva y se vio la necesidad de buscar otras posibilidades aprovechando las nuevas técnicas de imagen por MRI. En marzo de 2001 la eminente neuróloga Marjo van der Knaap y su equipo pudieron establecer una serie de criterios que probaban en un 90% de los casos la existencia de la enfermedad.[11] Se considera que la presencia de al menos 4 de los siguientes criterios dan un resultado diagnóstico positivo:
- Presencia de anormalidades extensas en la materia blanca con una preponderancia frontal o bien en cuanto a la extensión de estas anormalidades, en el grado de abultamiento, en el grado de cambio de señal o en el de pérdida de tejido (por atrofia o degeneración cística).
- Presencia de un borde periventricular de descenso de intensidad de señal en las imágenes potenciadas en T2 y elevación de la intensidad en imágenes potenciadas en T1.
- Anormalidades en los ganglios basales y tálamos, consistentes en una elevación en la intensidad de la señal y abultamiento o en atrofia y aumento o descenso de la intensidad de la señal en imágenes potenciadas en T2.
- Anormalidades en el tronco encefálico, en particular incluyendo el cerebro medio y la médula.
- Aumento de contraste que implica uno o más de las siguientes estructuras: borde ventricular, cerco de tejido periventricular, sustancia blanca de los lóbulos frontales, quiasma óptico, fórnix, ganglios basales, tálamo, núcleo dentado y estructuras troncoencefálicas.
Referencias
- ↑ Alexander WS (1949) Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain 72:373–381 PMID 15409268
- ↑ Schwankhaus JD, Parisi JE, Gulledge WR, Chin L, Currier RD (1995) Hereditary adult-onset Alexander's disease with palatal myoclonus, spastic paraparesis, and cerebellar ataxia. Neurology 45:2266–2271 PMID 8848205
- ↑ [http://ghr.nlm.nih.gov/condition=alexanderdisease Enfermedad de Alexander en Genetics Home Reference
- ↑ Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A (2001) Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nature Genet 27:117–120 PMID 11138011
- ↑ Li, R.; Johnson, A. B.; Salomons, G. S.; van der Knapp, M. S.; Rodriguez, D.; Boespflug-Tanguy, O.; Gorospe, J. R.; Goldman, J. E.; Messing, A.; Brenner, M. : Propensity for paternal inheritance of de novo mutations in Alexander disease. Hum. Genet. 119: 137-144, 2006. PMID 16365765
- ↑ RODRIGUEZ D y otros: Infantile Alexander disease: spectrum of GFAP mutations and genotype-phenotype correlation Am J Hum Genet. 2001 Nov;69(5):1134-40 PMID 11567214
- ↑ MEINS, M y otros: Novel mutations in exon 6 of the GFAP gene affect a highly conserved if motif in the rod domain 2B and are associated with early onset infantile Alexander disease. Neuropediatrics. 2007 Jun;38(3):143-7 PMID 17985264
- ↑ Quinlan RA, Brenner M, Goldman JE y Messing A: GFAP and its role in Alexander disease. Exp Cell Res. 2007 Jun 10;313(10):2077-87 PMID 17498694
- ↑ JOHNSON, AB: Alexander disease: a review and the gene. Int J Dev Neurosci. 2002 Jun-Aug;20(3-5):391-4 PMID 12175878
- ↑ Fawcet, D y Jensh, RP: Compendio de Histología. McGraw-Hill-Interamericana,1999 pgs 123-124 ISBN 84-486-0264-1
- ↑ a b van der Knaap, MS y otros: Alexander Disease:Diagnosis with MR Imaging. AJNR Am J Neuroradiol 22:541-552, marzo de 2001
- ↑ Russo LS Jr, Aron A, Anderson PJ: Alexander's disease: a report and reappraisal. Neurology. 1976 Jul;26(7):607-14 PMID 180453
- ↑ Ni Q, Johns GS, Manepalli A, Martin DS, Geller TJ:Infantile Alexander's disease: serial neuroradiologic findings. J Child Neurol. 2002 Jun;17(6):463-6 PMID 12174972
- ↑ Diana Rodríguez sobre la enfermedad de Alexander en Orphanet
Bibliografía
- Lazzarini, RA: Myelin Biology and Disorders. Academic Press, 2004 páginas 851 y stes. ISBN 0-12-439510-4
- The Official Parent's Sourcebook on Alexander Disease: Updated Directory for the Internet Age. Icon Health Publications, edición revisada (Diciembre de 2003) ISBN 0-597-83776-7 . Manual de referencia en investigación, diagnosis y consejos para padres y cuidadores.
- Parker, PM: Alexander Disease - A Bibliography and Dictionary for Physicians, Patients, and Genome Researchers. ICON Group International, Inc. (19 de julio de 2007) ISBN 0-497-11320-1.
Enlaces externos
Categorías:- Enfermedades raras
- Enfermedades neurológicas
- Enfermedades pediátricas
- Leucodistrofias
Wikimedia foundation. 2010.