- Síndrome X frágil
-
Síndrome X frágil 
Localización del gen FMR-1.Clasificación y recursos externos CIE-10 Q99.2 CIE-9 759.83 OMIM 309550 DiseasesDB 4973 eMedicine ped/800 MeSH D005600 Sinónimos - Retraso Mental ligado al Cromosoma X y Macroorquidia
- Síndrome de Martin Bell
- Síndrome del Marcador X
 Aviso médico
Aviso médico El síndrome del X frágil (SXF), también conocido como síndrome de Martin-Bell, es un trastorno hereditario que ocasiona retraso mental, pudiendo ser éste desde moderado a severo, y siendo la segunda causa genética del mismo, sólo superada por el síndrome de Down.
Afecta tanto a varones como a mujeres, si bien hay diferencias en las manifestaciones y en la incidencia del mismo. En varones, la incidencia es de 1 de cada 1.200, mientras que en mujeres es de 1 de cada 2.500, estando esta diferencia entre sexos estrechamente relacionada con la causa genética del síndrome.
La causa genética del síndrome es un tipo de mutación conocido como expansión de repeticiones de trinucleótidos, que supone el incremento en la descendencia del número de repeticiones de tres bases del ADN. Este tipo de mutación está asociado con el fenómeno de la anticipación, que se manifiesta como un aumento de la gravedad de los síntomas en sucesivas generaciones.
La mutación que origina el síndrome afecta a una región del cromosoma X en la que se sitúa el gen FMR-1. La expansión del trinucleótido tiene lugar en la región reguladora del gen, siendo este trinucleótido CGG (Citosina-Guanina-Guanina). Cuando el número de repeticiones supera el valor umbral de 230 repeticiones se produce la metilación del gen y, por tanto, éste pierde su función, produciendo así el síndrome del X frágil.
El producto de este gen, la proteína fmr1, puede encontrarse tanto en el núcleo como en el citoplasma, y a pesar de que su función es aún poco conocida, se ha visto que presenta la capacidad de unirse a determinados ARN mensajeros, por lo que dicha proteína podría estar implicada en el transporte de estos desde el núcleo hasta el citoplasma para su traducción.
Contenido
Historia
En 1943, Martin y Bell descubrieron un tipo de retraso mental hereditario ligado al X, que hoy conocemos como síndrome del X frágil. Ello ya se percataron de ciertas peculiaridades de los rasgos faciales de los pacientes y mencionaron que uno de los pacientes presentaba cara alargada y cejas prominentes.
En 1969, Lubs estudió una familia en la que cuatro varones de tres generaciones diferentes presentaban retraso mental. Los estudios citogenéticos de las muestras de estos pacientes revelaron una constricción inusual en el brazo largo del cromosoma X en el 10-33% de las células en cultivo. En un estudio posterior de la misma familia, Lubs y col., en 1984, describieron rasgos faciales inusuales en los miembros de esta familia que presentaban la afección: caras alargadas, orejas largas con inserción más baja de lo habitual, rasgos faciales asimétricos y cejas prominentes.
También en 1969, Opitz y col. emplearon el término "síndrome de Martin-Bell" para referirse a un caso de retraso mental familiar con características de dicho síndrome. En aquel entonces, nadie había relacionado el síndrome de Martin-Bell con el síndrome del X frágil de Lub.
En 1981, Richards y col. demostraron que ambos síndromes eran en realidad el mismo trastorno. Para ello, estudiaron a la misma familia que habían descrito Martin y Bell y utilizando la técnica de cultivo empleada por Lubs, observaron que todos los varones afectados presentaban el sitio frágil del cromosoma X en el 5-17% de sus células en cultivo.
En 1991, Verkerk y col. describieron un gen asociado al trastorno: el gen FMR-1 1 (acrónimo inglés de Fragile X linked Mental Retardation type 1; retraso mental ligado al X de tipo 1). Este descubrimiento ha traído consigo grandes mejoras en el diagnóstico prenatal y en la identificación de personas afectadas y en el rango de premutación.
Origen del nombre
El nombre del síndrome puede, de entrada, llevarnos a error. En los cromosomas de los pacientes que padecen este trastorno no hay una rotura del cromosoma X, ni siquiera hay un sitio frágil real en el mismo. X frágil hace alusión a una anomalía cromosómica estructural que se detecta en el brazo largo del cromosoma X en algunas células procedentes del paciente bajo ciertas condiciones de cultivo y que, debido a la manipulación de la muestra, puede romperse a nivel de esta anomalía, dando lugar a dos fragmentos cromosómicos. Es decir, el sitio frágil es fruto de la técnica y no se encuentra in vivo, sino sólo in vitro. Por tanto, no puede ser la causa de la enfermedad. Sin embargo, esta técnica de cultivo que permite observar la constricción secundaria del X frágil ha sido el críterio clásico de diagnóstico del trastorno, dado que gracias a ella podemos distinguir afectados de no afectados.
Genética
El hallazgo de los sitios frágiles contribuyó al descubrimiento de un nuevo tipo de mutación: la expansión de repeticiones de trinucleótidos; aunque no todas las mutaciones de este tipo producen sitios frágiles.
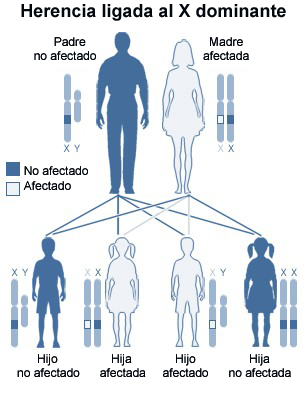
La herencia de esta mutación es de tipo dominante ligada al X, aunque no responde a las reglas usuales de dicha herencia, dado que hay portadores varones normales y mujeres portadoras no afectadas que dejarán su impronta (necesaria para la amplificación) e individuos afectados por el síndrome (mayoritariamente varones) entre la progenie de estas últimas.
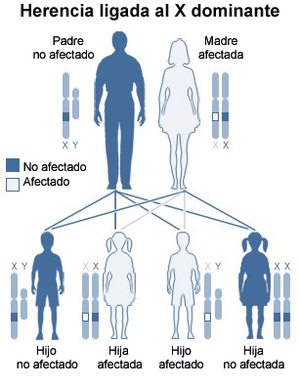 Diagrama de herencia ligada al cromosoma X dominante (madre afectada). El SXF presenta una mayor complejidad en su transmisión, ya que se produce la expansión de trinucleótido CGG.
Diagrama de herencia ligada al cromosoma X dominante (madre afectada). El SXF presenta una mayor complejidad en su transmisión, ya que se produce la expansión de trinucleótido CGG.
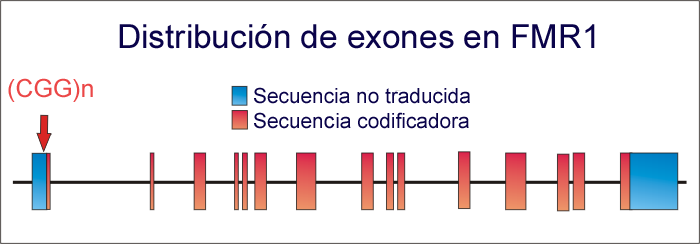
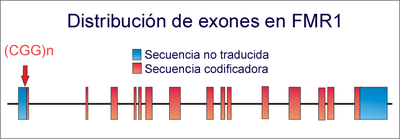 Distribución de exones en el gen FMR-1 y posición de las repeticiones del trinucleótido CGG (señalada por flecha). Cuando se supera el valor umbral, se produce el SXF.
Distribución de exones en el gen FMR-1 y posición de las repeticiones del trinucleótido CGG (señalada por flecha). Cuando se supera el valor umbral, se produce el SXF.Este síndrome presenta un fenómeno de anticipación: aumenta la penetrancia y la expresividad del trastorno a medida que transcurren las generaciones. Esto es debido al aumento del número de repeticiones de trinucleótidos CGG en el gen FMR-1.
El origen del síndrome del X frágil está en la inactivación de la transcripción de dicho gen. Esta inactivación se debe a la metilación del gen y ocurre cuando el número de repeticiones supera un valor umbral a partir del cual las enzimas metiladoras pueden llevar a cabo su función sobre dicho gen.
Al analizar mediante técnicas moleculares el ADN de pacientes del X frágil se observó que presentaban largas secuencias con cientos e incluso miles de repeticiones del trinucleótido CGG. Estas repeticiones se encuentran en una región no traducida (SANT) anterior al primer exón 1 del gen FMR-1, localizado en el sitio FRAXA, en la región Xq27.3. En personas no afectadas el número de repeticiones en esta región constituye un polimorfismo, siendo habituales valores de entre 5 y 55 repeticiones. La mutación consiste, por tanto, en la amplificación del número de repeticiones de triplete CGG.
Esta mutación no se expresa inmediatamente sino que evoluciona a lo largo de las generaciones, aumentando el número de repeticiones. Al principio atraviesa una etapa denominada "premutación" (entre 55 y 230 repeticiones), en la que no se expresa la síntomatología o está es leve, presentando solamente algunos de los síntomas y con menor severidad. Cuando el número de repeticiones supera el umbral de 230 repeticiones, se manifiesta el síndrome del X frágil, siendo frecuentes valores entre 230 y 1.000 o incluso superiores.
Las mujeres portadoras de una premutación corren el riesgo de tener hijos con el síndrome, siendo más probable cuanto mayor sea el número de repeticiones. A la hora de calcular la probabilidad de tener un descendiente afectado es importante considerar interrupciones de la repeticiones CGG por otras secuencias, dado que éstas se consideran preventivas de la expansión. Por ello es importante el análisis de la secuencia de la región Xq27.3 en las familias en las que se ha diagnosticado algún caso. En las familias en las que ha habido un caso de X frágil, alrededor del 10% de los varones normales portan la premutación. Todas las hijas de estos portadores heredarán la premutación, las cuales serán normales, pero sus descendientes varones tienen una alta probabilidad de sufrir el síndrome, debido a que durante la ovogénesis la madre deja su impronta génica en esta región cromosómica, la cual facilita la amplificación durante el desarrollo embrionario temprano de sus hijos, entre el 5º y el 20º día de vida, si bien este es un tema muy debatido y se ha planteado que la amplificación puede producirse durante la meiosis femenina.
Cuando el número de repeticiones supera el umbral, la secuencia es metilada por enzimas, extendiendose esta metilación a la isla CpG en la región reguladora del gen FMR-1. La transcripción se inhibe[1] [2] y como consecuencia se origina el síndrome.[3]
Se ha comprobado que es la inhibición de este gen la responsable del trastorno, ya que estudiando otros tipos de mutaciones génicas en el mismo,[4] se ha observado que éstas también producen el síndrome, aunque cabe destacar que son mucho más infrecuentes que la amplificación.
Los individuos que son citogenéticamente positivos por el sitio frágil en Xq27.3, pero son negativos por la expansión de CGG (lo cual está asociado con la mutación FRAXA), pueden tener una mutación más distal, incluyendo FRAXE o FRAXF.[5]
El descubrimiento del gen FMR-1 supuso un esfuerzo internacional que involucró a los laboratorios de Stephen Warren en Atlanta, David Nelson en Baylor, y de Ben Oostra en Holanda. Fue descrito por Verkerk y col., en 1991. Se expresa activamente en las espermatogonias, en las neuronas del hipocampo y del cerebelo y en muchos otros tipos celulares. Su producto, la proteína FMR-1, se localiza en el citoplasma y su función es poco conocida, aunque se ha comprobado que posee la capacidad de unirse al ARN, regulando la traducción de aproximadamente el 4% de éstos. Se piensa que esta proteína puede ser clave en la regulación de los cambios estructurales neuronales y en la maduración mediante la estimulación ambiental, particularmente en la selección de las conexiones neuronales.[6]
Consejo genético
Al igual que en cualquier otra enfermedad hereditaria, el consejo genético es importante en las familias con casos de síndrome del X frágil. Se debe asesorar a los individuos para que escojan libremente pero conscientes del riesgo que corren, tratando de estimar la probabilidad de que una pareja tenga un hijo afectado por el trastorno. También debe brindarseles información acerca de la enfermedad y su tratamiento.
Fenotipo y síntomas clínicos
Rasgos y síntomas 
Rasgos y síntomas: cara alargada, frente prominente, mentón pronunciado, grandes orejas.
- Retraso mental.
- Hiperactividad.
- Problemas de atención.
- Contacto visual escaso.
- Habla reiterativa.
- Articulaciones hiperextensibles.
- Testículos grandes.
- Orejas prominentes.
- Bajo tono muscular.[7]
Las características principales de este síndrome, si bien individualmente no son exclusivas de este trastorno, han de tenerse muy en cuenta en personas con autismo, retraso mental o problemas con el aprendizaje. La posesión de varios de estos rasgos y síntomas por parte de una persona puede hacer sospechar la presencia del síndrome y debe optarse por realizar el diagnóstico oportuno, dado que se trata de una enfermedad familiar.
Dichos rasgos son retraso mental profundo, especialmente en varones, aumento del volumen testicular por encima de 30mL (macroorquidismo) y peculiaridades faciales y del tejido conectivo.
Debido a una reducción de la distancia intercigomática la forma del rostro es más alargada de lo habitual. Otras características faciales y craneanas típicas, si bien no tienen porque encontrarse en todos los pacientes, son macrocefalia, rostro áspero, frente amplia, cejas prominentes y orejas largas, a menudo con inserción baja. En cuanto al macroorquidismo, en la mayoría de los casos no se manifiesta hasta pasada la pubertad, si bien se han detectado algunos casos de macroorquidismo congenito. En lo referente al tejido conectivo, el paciente puede presentar escoliosis, articulaciones laxas y pies planos. Otros rasgos físicos son pecho excavado, válvula mitral prolapsa, leve dilatación de la aorta ascendente y heterotopía periventricular. Los cambios neuroanatómicos en el cerebro de individuos con el síndrome de X frágil incluyen un agrandamiento del núcleo caudado, del hipocampo, y ventrículos laterales. El vermis cerebeloso es más pequeño de lo normal. El tamaño del cerebelo está correlacionado con el nivel cognitivo, incluyendo la función ejecutiva.[8]
En lo referente a rasgos psíquicos, el más significativo es el retraso mental, siendo más acentuado en los varones y generalmente profundo (aunque en algunos casos puede ser moderado), mientras que en las mujeres suele ser leve. El CI de los afectados varones se situá entre 35 y 45, mientras que en el caso de las mujeres afectadas el CI está menos afectado, situandose entre 60 y 80. Además, éstas presentan signos somáticos más leves. Esto es debido al mosaicismo que presentan las mujeres, debido a la heterocromatinización al azar de uno de sus cromosomas X en cada célula durante el desarrollo embrionario. Aproximadamente el 70% de las mujeres con la mutación completa tienen un déficit cognitivo en el límite o en el rango de retraso mental,[9] mientras que aproximadamente el 85% de los varones con la mutación completa son retrasados mentales.[10] Los varones que presentan un menor retraso e incluso carecen de él, usualmente presentan mosaicismo, es decir, algunas células poseen premutación y otras mutación completa o no presentan metilación a pesar de poseer la mutación completa. También son frecuentes los movimientos estereotipados de la cabeza y las manos y las manifestaciones psiquiátricas y de personalidad, así como la hiperactividad y el autismo. Generalmente, los pacientes de síndrome del X frágil presentan pobre o nulo contacto visual y son habituales los periodos de agresividad alternados con periodos de notable timidez. También son habituales las dificultades en el uso del lenguaje y en el aprendizaje, especialmente de las matemáticas, y los problemas de integración sensorial debidos a la dificultad para comprender los estímulos (visuales, auditivos o táctiles), así como el rechazo sistemático a nuevos estímulos.
Ataxia y tremor asociados al X frágil (FXTAS)
Es un trastorno neurodegenerativo tardío asociado con problemas con los movimientos, de memoria y del sistema nervioso autónomo. El FXTAS puede presentar muchos de los síntomas de la atrofia multisistémica y con frecuencia incluye parkinsonismo, disautonomía, neuropatía periférica, y demencia.
Este trastorno es debido a una extensión de trinucleótidos CGG en el gen FMR-1, en un rango de entre 55 y 230 repeticiones, es decir, en el rango de premutación del retraso mental ligado al X frágil. A pesar de que involucra a este gen, es un trastorno clínico muy diferente.
Se presenta con mayor frecuencia en los hombres, pero también puede presentarse en mujeres. No existe cura para el FXTAS, pero algunos de sus síntomas pueden mejorarse con medicación.
Diagnóstico
Cuando un individuo con retraso mental o autismo presenta algunos de los rasgos característicos de los mencionados con anterioridad, se sospecha que puede estar afectado por el síndrome. Pero no basta con detectar síntomas somáticos y retraso mental para dar un diagnóstico positivo del trastorno, sino que hay que recurrir al diagnóstico genético para que este sea definitivo.
Clásicamente, el diagnóstico definitivo de la enfermedad se establecía citogenéticamente, por la expresión del sitio frágil en ciertas condiciones de cultivo. Un sitio frágil es una región o banda cromosómica que aparece como una interrupción no coloreada y que puede romperse mientras se trabaja con la muestra, dando lugar a fragmentos cromosómicos de tamaño definido. Pero como ya se dijo, el sitio frágil no se expresa in vivo. Para que se haga patente es necesario cultivar las células del paciente (linfocitos o fibroblastos) en un medio pobre en ácido fólico y desoxitimidintrifosfato (dTTP) durante, al menos, un ciclo celular. Es decir, es necesario que ocurra la etapa de síntesis de ADN (S) al menos una vez para que esta constricción se manifieste. Que aparezca o no en una célula de una persona afecta es un hecho probabilístico y su patencia suele darse en el 5%-20% de las células, por lo que es necesario observar muchas células antes de poder dar un diagnóstico citogenéticamente negativo. En cambio, basta con encontrar una célula o pocas células con la constricción para dar un diagnóstico citogenéticamente positivo.
A microscopía óptica y con un bandeo cromosómico se puede apreciar una región alargada y condensada próxima al extremo del brazo largo del cromosoma X, entre la banda q27 y q28, si bien sabemos que está exactamente en q27.3. Al microscopio electrónico tiene el aspecto de una constricción secundaria tras la cual queda un gran satélite.
El primer sitio frágil del cromosoma X que se detectó fue el sitio FRAXA, que es el que se ha descrito hasta ahora, siendo además el más abundante. Este sitio frágil afecta al gen FMR-1. Con posterioridad, se detectaron otros sitios frágiles menos frecuentes también en el cromosoma X, siendo el más importante de ellos el sitio FRAXE, dado que presenta asociación con retraso mental leve y afecta al gen FMR-2, cuya función no está muy clara. Está localizado en Xq28. En cuanto a los otros sitios, FRAXD y FRAXF están poco estudiados y se desconoce o se conoce poco acerca del fenotipo asociado. Se sabe que FRAXD está en Xq27.2, muy próximo al sitio FRAXA, y que su constricción es inducible por altas dosis de afidicolina, como ya demostró Sutherland en 1989. Tan sólo se detecta en el 1%-2% de los pacientes, por lo que no es muy significativo, y además puede detectarse por el procedimiento habitual. El FRAXF aparece en personas sin afección, como una lesión cromosómica en Xq26. No se conoce mucho más acerca de este sitio frágil.
Desde los años 80 se han descubierto descubierto, al menos, otras doce constricciones secundarias heredables en otros cromosomas, pero ninguna de ellas está asociada a un fenotipo en particular.
En 1981 y 1982, Tommerup y col. y Jacobs y col. demostraron que la inhibición farmacológica de la timidilato sintetasa (TYMS) es efectiva induciendo la marca del X frágil en células en cultivo.
En 1991, Griffiths y Strachan describieron una técnica que permite visualizar el sitio frágil y hacer un bandeo prometafásico en el mismo espécimen.
En la actualidad, se prefieren técnicas moleculares para el diagnóstico definitivo, dado que conocer el número de repeticiones en la secuencia puede ser muy útil para estudiar la herencia de la enfermedad dentro de una familia, ya que permite estudiar individuos no afectados no portadores, individuos no afectados portadores e individuos afectados portadores. En estos últimos, además permite estudiar el grado de metilación, decisivo en la manifestación del síndrome.
Otra técnica de diagnóstico consiste en el uso de enzimas de restricción y posterior electroforesis de los fragmentos con el fin de hallar bandas de longitud anormal. Combinando enzima sensibles a la metilación con otras que no lo son pero que tienen la misma secuencia de reconocimiento y patrón de corte se pueden detectar metilación anormal en el sitio frágil tanto en varones afectados como en mujeres portadoras. Algunos varones afectados aparentan ser mosaicos, con la coexistencia de un largo fragmento metilado y uno corto normal sin metilar.
El empleo del Southern blot con digestiones de EcoRI y EagI es un sencillo test para distinguir el genotipo normal, la premutación y la mutación completa.
También puede usarse la prueba pfx3 o una PCR seguida de una secuenciación para conocer el número exacto de repeticiones, especialmente si éstas superan las 130.
Una técnica que resulta intersante por ser poco invasiva es el empleo de anticuerpos monoclonales de ratón contra la proteína FMR-1 en un frotis sanguíneo del paciente. Es muy poco invasiva, ya que tan sólo requiere una o dos gotas de sangre. Una adaptación de esta misma prueba se ha empleado para hacer el diagnóstico con raíces capilares en lugar de con muestras sanguíneas. Esto puede ser de utilidad si pensamos que algunos pacientes poseen trastornos de la personalidad que se manifiestan con frecuencia en forma de agresividad. Un manera sencilla de solventar las posibles molestias de optener una muestra sanguínea es recoger capilares desprendidos en las sabanas, en la almohada o mediante el uso de un peine o cepillo para realizar el diagnóstico.
En contra posición, una técnica invasiva pero que puede emplearse si no se quiere recurrir a técnicas moleculares consiste en el análisis de neuroblastos olfatorios, porque son neuronas accesibles que pueden regenerarse y que están estrechamente unidas al cerebro.
MacKenzie y col., en 2006, estudiaron la posibilidad de utilizar derivados del ectodermo para el diagnóstico del síndrome.
Diagnóstico prenatal
En el caso familias con antecedentes del síndrome, el diagnóstico prenatal puede contribuir a mejorar la calidad de vida de los descendientes, especialmente de mujeres portadoras de la premutación.
Aplicado a un embrión en gestación en etapas tempranas del desarrollo puede servir para tomar la decisión de abortar o no en el caso de que se detecte que éste posee la mutación completa y que se sospeche que con alta probabilidad va a sufrir retraso mental severo.
También puede emplearse para conocer si embriones en etapas más tardias del desarrollo tiene alta probabilidad de sufrir el síndrome y así adecuar el entorno en el que se va a desarrollar el niño y comenzar con el tratamiento a edades tempranas, con el fin de mejorar las capacidades cognitivas.
Para llevar a cabo este diagnóstico pueden emplearse cultivos de amniocitos o la secuenciación a partir de vellosidades coriónicas. También puede utilizarse la técnica pfxa3 para tratar de detectar una banda anormal de 2,3kb. En embriones más desarrollados puede extraerse una muestra de sangre para realizar un frotis sanguíneo junto con anticuerpos monoclonales de ratón contra la proteína FMR-1.
El diagnóstico prenatal también se puede emplear en mujeres con la premutación que hallan empleado la fecundación in vitro. Antes de la implantación de los embriones, se pueden utilizar diversos métodos de diagnóstico molecular con la intención de seleccionar embriones sanos.
Previo al diagnóstico prenatal se encuentra el consejo genético, basado en estudios genéticos y fenotípico de los padres y sus familiares.
Tratamiento
El tratamiento de pacientes con el síndrome de X frágil es bastante complejo y su efectividad está bastante limitada. Involucra a múltiples profesionales: especialistas en educación especial, terapeutas ocupacionales, psicólogos, logopedas, pedagogas y médicos. El asesoramiento genético enfocado a las familias implicadas es esencial, donde juegan un papel fundamental el consejo genético. El espectro de compromiso con el tratamiento es un asunto analizados en detalle entre el médico y la familia.[11] Los niños afectados por el síndrome suelen requerir terapia del lenguaje y terapia ocupacional,[12] pudiendo mediarse éstas a través del centro educativo del paciente. Los varones en particular tienen problemas significativos de integración sensorial. Técnicas conductuales junto con terapias de coordinación motora fina y gruesa pueden apaciguar el estado anímico del paciente. Los trastornos de comportamiento severo, requieren la intervención de pedagogos y psicólogos que enseñen a la familia técnicas de comportamiento.
El uso de medicación psicotrópica es una herramienta útil en muchos casos. Mejorar la concentración y disminuir la agresividad, en el caso de que esté presente, son los objetivos principales en la niñez temprana. Entre los afectados por este síndrome, y particularmente en niños de edad preescolar, las medicaciones estimulantes, como el metilfenidato, se asocian a menudo con un incremento de la irritabilidad. La clonidina, que tiene una acción apaciguante, ayuda a controlar los síntomas de hiperactividad y agresividad en la mayoría de los niños con X frágil. Hay que realizar un cuidadoso seguimiento con electrocardiogramas periódicos si se emplea algún tipo de medicación psicotrópica.
En niños en edad escolar, los estimulantes (metilfenidato, dextroanfetamina y Adderall) son eficaces en aproximadamente el 60% de los casos. En lo que respecta a los agentes anticonvulsivos, como carbamazepina o ácido valproico, son la principal elección ante cuadros de significativa inestabilidad emocional.[13] Cuando el paciente padece ansiedad, desasosiego o agresividad, también se utilizan Inhibidores de la Recaptación de Serotonina (IRS), como la fluoxetina, la sertralina, la fluvoxamina o el citalopram.
Se están poniendo a prueba diferentes moléculas de acción neurotónica, incluyendo agonistas de los receptores AMPA y antagonistas selectivos de los receptores glutamatérgicos, que podrían tener gran aplicación en el tratamiento farmacológico del síndrome.
Existe un grupo de investigación interdisciplinar dirigido por la doctora Yolanda de Diego Otero de la Fundación IMABIS, en Málaga, que avanza en el desarrollo de un nuevo tratamiento para el Síndrome X Frágil. La Agencia Española del Medicamento ha aprobado el ensayo clinico, financiado principalmente por el Ministerio de Sanidad y Política Social, que se está desarrollando actualmente para comprobar la efectividad de compuestos antioxidantes en la mejora de las alteraciones de comportamiento y aprendizaje de los afectados por el Síndrome, descubrimiento que ha sido objeto de protección por una patente de invención.
Los últimos resultados de sus investigaciones han permitido describir una nueva diana terapéutica, para diseñar tratamientos específicos para el Síndrome X frágil e investigar sus efectos sobre la enfermedad. Dos de las más prestigiosas revistas científicas del campo de la neurociencia “Neuropsychopharmacology” y “Journal of Pineal Research”, han publicado los resultados más recientes de la investigación, en dos artículos complementarios donde se describe por primera vez que existen compuestos que controlan parte de la sintomatología, actuando sobre la eliminación y el control de la producción de radicales libres, el mecanismo bioquímico alterado en el cerebro del ratón afectado con el Síndrome, como previamente ya había descrito este mismo grupo de investigación. Los compuestos reguladores del estrés oxidativo, contrarrestan la producción de radicales libres y mejoran el comportamiento y el aprendizaje de los ratones afectados por el Síndrome X frágil.
Bibliografía
En castellano
- "Síndrome X Frágil. Libro de consultas para familias y profesionales." Dir: MªIsabel Tejada (Pta.Asoc.GIRMOGEN). Edita: Real Patronato sobre Discapacidad. Madrid, 2006. NIPO: 214-08-002-0 Copia gratuita en PDF
- " Síndrome X Frágil y discapacidad mental hereditaria". Autores: Yolanda de Diego Otero y col. Edita: Ministerio de Sanidad y consumo. Madrid 1999. NIPO: 351-99-005-2.
- Thompson, Margaret W., McInnes, Roderick R., Willard, Huntington F. "Thompson & Thompson. Genética médica". 7ª edición. Ed. Elsevier Masson.
- Jorde, Lynn B., Carey, John C., Bamshad, Michael J. "Medical genetics". 2ª edición en español. Ed. Harcourt.
- Solari, Alberto J. "Genética humana: fundamentos y aplicaciones en medicina". 3ª edición. Ed. Panamericana.
En inglés
- Abitbol M, et al. (1993): "Nucleus basalis magnocellularis and hippocampus are the major sites of FMR1 expression in the human fetal brain". Nature Genetics, 4:147-53. PMID 8348153
- Ashley CT Jr., et al. (1993): "FMR1 protein: conserved RNP family domains and selective RNA binding". Science, vol. 262:563-5. PMID 7692601
- Bakker CE, et al. (1994): "FMR1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium". Cell, 78, 23-33. PMID 8033209
- Baumgardner TL, et al. (1995): "Specification of the neurobehavioral phenotype in males with fragile X syndrome". Pediatrics, 95:744-52.
- Hagerman RJ, et al. (1994): "High functioning fragile X males: Demonstration of an unmethylated fully expanded FMR1 mutation associated with protein expression". Am J Med Genetics, 51:298-308.
- Verkerk AJ, et al. (1991): "Identification of a gene (FMR1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome". Cell, 65, 905-14.
- Lecturas adicionales (actualizaciones)
- Bakker CE, et al. "Understanding fragile X syndrome: insights from animal models". Cytogenet Genome Res. 2003;100(1-4):111-23. Revisión. PMID 14526171
- Koekkoek SK, et al. "Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome". Neuron. 2005 ag 4;47(3):339-52. PMID 16055059
Referencias
- ↑ Pieretti M, et al (1991). "Absence of expression of the FMR1 gene in fragile X syndrome." Cell, 66:817-22.
- ↑ Imbert G, et al (1998). "FMR1 and mutations in fragile X syndrome: Molecular biology, biochemistry, and genetics." En Well RD, et al (ed): Genetic Instabilities and Hereditary Neurological Diseases. San Diego: Academic Press. p15-25.
- ↑ Oostra BA (1996). "FMR1 Protein Studies and Animal Models for Fragile X Syndrome." En Fragile X Syndrome: Diagnosis, Treatment and Research, ya citado.
- ↑ De Boulle K, et al (1993). "A point mutation in the FMR1 gene associated with fragile X mental retardation." Nature Genetics, Vol. 3, pg 31-5.
- ↑ Flynn GA, et al (1993). "Identification of the FRAX-E fragile site in two families ascertained for X linked mental retardation." Journal of Medical Genetics, 30:97-100.
- ↑ Comery TA, et al (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits Proc. Natl. Acad. Sci. USA, 94:5401-4. PMID 9144249
- ↑ Goldstein, Sam, and Cecil R. Reynolds. Handbook of neurodevelopmental and genetic disorders in children. Guilford P, 1999.
- ↑ Mostofsky WH, et al (1998). "Decreased cerebellar posterior vermis size in fragile X syndrome." Am Acad Neurol. 50:121-30.
- ↑ deVries BBA, et al (1996). "Mental Status of females with an FMR1 gene full mutation." Am J Hum Genet, 58:1025-32.
- ↑ Hagerman RJ (1999). "Fragile X Syndrome." En Neurodevelopmental disorders: Diagnosis and Treatment. Nueva York: Oxford University Press.
- ↑ Cronister, A (1996). "Genetic Counseling." En Hagerman RJ, et al (ed): Fragile X Syndrome: Diagnosis, Treatment and Research, segunda edición. Baltimore: Johns Hopkins University Press.
- ↑ Scharfenaker S, et al (1996). "An Integrated Approach to Intervention." En Fragile X Syndrome: Diagnosis, Treatment and Research, ya citado.
- ↑ Hagerman RJ (1996). "Medical Follow-up and Pharmacotherapy." En Fragile X Syndrome: Diagnosis, Treatment and Research, ya citado.
Véase también
- Enfermedad rara
- Genética
- Genoma humano
- Genoma
- Prueba genética
- Síndrome de Down
- Síndrome de Prader-Willi
- Prolapso de la válvula mitral
Enlaces externos
- Federación Española del Síndrome X Frágil
- Foro de la Asociación Síndrome X Frágil de Madrid para orientar, asesorar, promocionar la investigación, colaborar en áreas educativas y centros de personas o profesionales dedicados al estudio del Síndrome X Frágil y sus afectados.
- En MedlinePlus puede encontrar más información sobre Síndrome X frágil
- En Medline hay más información sobre Síndrome X frágil (en inglés)
- www.sindromexfragil.com.ar Agrupación Síndrome X Frágil de Argentina.
- www.euskalnet.net/axfrav/ Asociación X Frágil del País Vasco.
- vi.vu/es/topics/sindrome-x-fragi Vivu portal médico.
- Fundación IMABIS Investigadores de la Fundación Imabis avanzan en el desarrollo de nuevos enfoques terapéuticos para el Síndrome X Frágil
- En inglés
- National Genome Research Institute
- FraXA.org The Fragile X Research Foundation.
- FragileX.org The National Fragile X Foundation.
- Stanford.edu Trinucleotide Repeat Disorders Part 9: Non-Polyglutamine Diseases: Descriptions of other diseases that involve codon repeat expansions (Septiembre 18, 2002).
- NIH.gov Fragile Site Mental Retardation 1 Gene; FMR1 (entrada OMIM para el SXF).
- GeneClinics.org "Fragile X Syndrome: FRAXA, FXS, Fragile X Mental Retardation, Marker X Syndrome, Martin-Bell Syndrome". Incluye: Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS), Robert A. Saul, MD, FACMG, Jack C. Tarleton, PhD, FACMG, GeneReview (Septiembre 13, 2004).
- Nature.com - From MRNP Trafficking to Spine Dysmorphogenesis: The Roots of Fragile X Syndrome, Claudia Bagni & William T. Greenough. Nature Reviews Neuroscience, 6, pp 376–387 (2005)
Categorías:- Síndromes
- Enfermedades hereditarias
- Enfermedades raras
- Anomalías estructurales cromosómicas

Wikimedia foundation. 2010.