- Síndrome de Prader-Willi
-
Síndrome de Prader-Willi 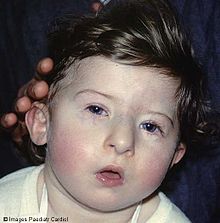
Paciente con el síndrome, mostrando las características faciales comunes como cara elongada, nariz prominente y surco nasolabial liso.Clasificación y recursos externos CIE-10 Q87.1 CIE-9 759.81 OMIM 176270 DiseasesDB 10481 MedlinePlus Información de salud en la enciclopedia MedlinePlus eMedicine ped/1880 MeSH D011218  Aviso médico
Aviso médico El síndrome de Prader-Willi (SPW) es una alteración genética descrita en el año 1956 por los doctores suizos Andrea Prader, Alexis Labhart y Heinrich Willi,[1] en nueve pacientes que presentaban un cuadro clínico de obesidad, talla baja, hipogonadismo, criptorquidia y alteraciones en el aprendizaje tras una etapa de hipotonía muscular pre- y posnatal, además de una discapacidad intelectual de leve a moderada.
La incidencia y frecuencia publicada es muy variable, aceptándose que entre 1 de cada 10.000 niños y 1 de cada 30.000 niños (en función de las poblaciones) nace con esta compleja alteración genética.[2] Considerada una enfermedad rara, parte de su complejidad se basa en el amplio rango de manifestaciones clínicas y en su variable grado de severidad.
Contenido
Síntomas
Se conoce como síndrome a un conjunto de síntomas y signos significativos, que aunque en determinadas ocasiones puede ser debido en exclusiva a una enfermedad, es por lo general de naturaleza compleja y con diversas causas y manifestaciones. La genética de la enfermedad es bastante compleja, y es un ejemplo clásico de lo que se conoce como impronta genética.
Este síndrome altera el funcionamiento del hipotálamo, una sección del diencéfalo cuyas funciones incluyen, entre otras, el control del apetito lo que provoca que carezcan de sensación de saciedad. La observación clínica y algunos trabajos de investigación, han demostrado una diferencia entre “sensación de hambre” y “falta de saciedad”[cita requerida]. Un error muy común es pensar que la búsqueda incesante de “comida” se debe a un “hambre excesiva”. La alimentación de las personas con SPW necesita estar supervisada constantemente, además de seguir una dieta estricta.
 Uno de los aspectos más importantes a tratar es el hábito alimenticio.
Uno de los aspectos más importantes a tratar es el hábito alimenticio.
Provoca asimismo deficiencia del tono muscular, un alto porcentaje de grasa en el organismo y falta de energía. Todas estas condiciones reducen las necesidades calóricas de los niños y adultos que tienen este síndrome a dos tercios de la necesidad calórica estándar.
Si bien el trastorno alimenticio es el síntoma más evidente y el que demanda más tiempo, además de su mayor riesgo vital, es sólo un aspecto de esta compleja dolencia. Al principio, los bebés que tienen este síndrome se alimentan de forma deficiente y no aumentan de peso, ya que la debilidad de su tono muscular reduce su capacidad de succión.
El síndrome de Prader-Willi también puede provocar crecimiento y maduración incompletos, facciones características, problemas del comportamiento, dificultades respiratorias, comportamiento obsesivo-compulsivo (como hurgarse en lesiones en la piel, pensamientos y acciones repetitivos y una fuerte necesidad de seguir una rutina), disfunciones en la temperatura corporal, resistencia al dolor, retraso en el desarrollo del aprendizaje y, en dos terceras partes de los casos, imposibilidad de vomitar. Alguno puede llegar a comerse cualquier cosa, y los medicamentos para inducir al vómito son ineficaces y pueden resultar tóxicos.
La hipopigmentación suele ser también una característica de estos pacientes.
Características PsicológicasEn general, las personas con SPW suelen sufrir alguna limitación cognitiva. Dentro de esta limitación, existen grandes diferencias interindividuales, siendo el cociente intelectual medio alrededor 70[cita requerida].
A nivel cognitivo, estas personas normalmente tienen buena memoria a largo plazo, buena organización perceptiva, buena habilidad para reconocer y evaluar relaciones espaciales, buena decodificación y comprensión lectora y un buen vocabulario expresivo. Sin embargo, suelen presentar: falta de procesamiento secuencial de la información, dificultades en la aritmética, pobre memoria a corto plazo (por ejemplo, les cuesta recordar cadenas de información, como pueden ser órdenes por lo que se tacha muchas veces al niño de desobediente), tienen dificultades de atención y concentración y habilidades motoras finas relacionadas con la planificación motriz, el tono o la fuerza.
ConductasLas personas con SPW suelen tener un patrón conductual específico en el que se observan fluctuaciones significativas según la edad. Los niños pequeños suelen ser alegres, afectuosos, complacientes y cooperadores hasta que sobre los 6 u 8 años se vuelven más rígidos, irritables y emocionalmente más inseguros. Así comienzan a tener conductas como engullir toda la comida disponible, impaciencia, ataques de ira, enfados, distracciones, problemas de comunicación e impulsividad, suelen ser manipuladores, mentirosos, hábiles, caprichosos, egocéntricos; con frecuencia muestran conductas autolesivas y tienen pocas habilidades interpersonales. Las habilidades de cooperación suelen estar más alteradas, aunque éstas mejoran con la edad.
En las actividades de la vida diaria, las personas con SPW se desenvuelven relativamente bien. Destacan especialmente en las habilidades domésticas de preparación de la comida (en los casos en los que se les permite y no les genera demasiado estrés) y en tareas de auto-ayuda.
Debido a que la familia supervisa permanentemente todo lo que tiene que ver con la comida, se desenvuelven en un área donde no tiene ningún control. En un intento por relacionarse o luchar con el mundo, se encierran en lo que saben (o creen saber), evitando la inclusión de cualquier información contradictoria, y cuando esto último sucede, se muestran ansiosos y agitados e incluso se esfuerzan aún más por adquirir un control interno mayor; pero no pueden, y en estos casos se dan conflictos emocionales.
Características del habla y del lenguaje en personas con SPW
El patrón de desarrollo del lenguaje en las personas con Síndrome de Prader Willi es el mismo que el de toda la población aunque el ritmo es más lento. De esta manera, sus primeras palabras suelen aparecer en torno a los dos años y medio y la producción verbal significativa a menudo es escasa antes de los cuatro años.
Algunas de las características más importantes que a nivel lingüístico presentan los sujetos con SPW son:
- Los errores más comunes son: las distorsiones, las omisiones, simplificaciones de fonemas y dificultades en la secuenciación de sílabas.
- Suelen tener un tono de voz inadecuado (bajo para las niñas y alto para los niños) y presentar hipernasalidad (debido a las dificultades de la función motora oral).
- Carecen de un amplio vocabulario.
- El pensamiento concreto y la falta de experiencias provocan dificultades en la generalización de conceptos.
- Dificultades en la compresión de oraciones negativas e interrogativas ya que exigen cierto nivel de abstracción y capacidad de ponerse en el lugar del otro.
- Suelen tener mayores problemas en las construcciones sintácticas que en las morfológicas.
- Tienen limitaciones para construir frases ya que esto exige creatividad y capacidad de organizar y combinar distintos elementos.
- La estructuración de la oración suele ser más lenta y las producciones suelen ser incorrectas e incompletas. Suele ser poco frecuente la utilización de nexos y de oraciones compuestas.
- El uso del lenguaje está muy relacionado con el propio nivel lingüístico de la persona, con el ambiente que le rodea y con la cantidad y calidad de las interacciones verbales de su experiencia.
- La falta de comprensión de los mensajes, les lleva muchas veces a mantener conversaciones sin sentido o a la inhibición y al desinterés comunicativos.
- El carácter sociable y tímido de estas personas es un aspecto favorecedor en el uso del lenguaje, aunque en las situaciones comunicativas se observan problemas relacionados con la proximidad física al hablar, el respeto del turno de palabras, la posición de escucha.
LectoescrituraEl éxito del aprendizaje de la lectoescritura dependerá de la expresión/comprensión oral alcanzando en los años procedentes así como el grado de afectación motora pero todas las personas deben enfrentarse a su aprendizaje. La decodificación suele ser mejor que la comprensión, por eso suelen ser niños que recuerdan muy poco de lo leído y tienen dificultades para referirlo en el orden correcto.
Genética
El síndrome de Prader-Willi es una enfermedad genética producida por la ausencia de la expresión de un alelo localizado en el brazo largo del cromosoma 15 de origen paterno (concretamente en la región 15q11-q13). Esta ausencia de expresión puede deberse a varias causas, y es por ello que la herencia de este síndrome es compleja. Es importante llamar aquí la atención sobre la semejanza génica de este síndrome con el de Angelman, en el que la enfermedad se desarrolla por la ausencia de expresión de varios alelos en el mismo locus, pero en este caso, de origen materno.
En individuos sanos, el cromosoma paterno expresa varios genes (SNRPN, NDN, MAGEL2, cluster snARN), e inhibe la del gen UBE3A. Lo hace porque no se encuentra metilada la diana de impronta génica. En cambio, el cromosoma materno sí tiene metilada esta diana, por lo que inhibe la expresión de los genes que se expresaban en el paterno y activa la de UBE3A. El efecto fundamental es que se deja de sintetizar una ribonucleoproteína. Esto es lo que ocurre en condiciones normales, en las que los óvulos y espermatozoides realizan el proceso de impronta génica correctamente. El síndrome de Prader-Willi se produce al faltar la expresión de los genes que un cromosoma paterno silvestre o sano expresaría. La falta de esta expresión puede ser debida a varias causas:
- Deleción o pérdida de la región 15q11-q13 del cromosoma de origen paterno. Se observa en el 70% de los pacientes.[3] El riesgo de recurrencia familiar es cercano al 1%
- Disomía uniparental materna, es decir, la presencia de dos cromosomas maternos en vez de uno paterno y otro materno.[4] Se observa en aproximadamente un 20% de los pacientes.[5] El riesgo de recurrencia no supera el 1%
- Defectos en la impronta de la diana que silencien los genes que deberían expresarse en el paterno. En este caso aunque la frecuencia es menor, si se presenta el riesgo de recurrencia puede llegar al 50%
Diagnóstico tradicional y diagnóstico molecular
Dado que no existe tratamiento para la cura de la enfermedad, el diagnóstico temprano para la correcta atención a los síntomas es muy importante.
A nivel molecular, los principales test aplicados en la clínica son:
- Análisis de metilación por PCR: Técnica[6] por la cual si el patrón de metilación hallado corresponde únicamente al materno, se confirma el diagnóstico, que puede ser de PWS asociado a deleciones, disomía uniparental o defectos en impronta.
Con respecto al diagnóstico no molecular, existe un consenso,[7] establecido por primera vez en el año 93 y que fue ratificado recientemente, en el año 2001.[8]Tradicionalmente se han considerado en el diagnóstico aspectos como la hipotonía, la corta estatura, la hiperfagia, la obesidad, el comportamiento (específicamente con trastornos del tipo obsesivo-compulsivo, el tamaño pequeño de manos y pies, el hipogonadismo y un retraso mental leve. Hoy en día el diagnóstico se confirma con test genéticos, y de hecho se recomienda el uso del test sobre los recién nacidos que presentan una hipotonía significativa.
Las razones básicas del interés en un diagnóstico temprano son:1ª “Conocer” el previsible proceso que presentará la persona. En ocasiones existen tratamientos paliativos y el diagnóstico posibilita conocer y anticiparse a las complicaciones que puede presentar la persona con SPW. El administrar la terapia adecuada en el momento oportuno mejorará el pronóstico de la enfermedad.
2ª Dirigirse a las diferentes Asociaciones, para que se les dé una información específica de cómo se desarrolla la vida de otras personas que presentan el mismo síndrome y a su vez, le sirva de apoyo emocional a la familia. También, las asociaciones pueden dirigir a la familia a los profesionales, investigadores, que traten este síndrome.
3ª Ofrecer un consejo genético, por el cual, la persona o la familia con riesgo de este trastorno genético, sean informadas de las consecuencias de dicho trastorno, de la probabilidad de tenerlo y de la forma en que se pueda evitar o mejorar.
Escolarización e iniciativas para el aula
Para la escolarización de alumnos con SPW existen varias alternativas, siendo la más usual y adecuada la escolarización en un centro ordinario, para favorecer así su integración social. Todo ello dependerá las necesidades educativas de cada alumno así como de los recursos que los centros ordinarios sean capaces de ofrecer para satisfacer dichas necesidades.
Para los alumnos con SPW es muy importante la Atención Temprana, ya que tienen unas altas capacidades en el aprendizaje y cuanto antes comience, el desarrollo de sus capacidades mediante la intervención educativa, mejor será su desarrollo y calidad de vida futura.
Es conveniente realizar periódicamente revisiones en el dictamen de escolarización ya que las necesidades de estos alumnos varían con el tiempo y por lo tanto los recursos y apoyos específicos deben hacerlo también.
A la hora de la escolarización del alumnado con SPW se deberá tener en cuenta que para que el alumno desarrolle al máximo sus capacidades hay que procurar que el alumno reciba el proceso de enseñanza-aprendizaje en un ambiente lo más normalizado posible, permitiéndole así la integración en la sociedad y optimizando su calidad de vida.
Todos los profesionales que trabajen con el alumno deberán conocer tanto las necesidades educativas que presentan como las características médicas que presenta ya que esto afecta a su rendimiento escolar.
Es imprescindible la colaboración de todos los profesionales que atienden al alumno, así como el contacto con la familia, que desempeñan un papel principal en la educación de sus hijos.
Estrategias para el aulaDe entre todas las características que los alumnos con SPW pueden presentar, existen algunas de ellas que los profesores deben tener más en cuenta a la hora de trabajar en el aula. Ya que afectan al proceso de enseñanza-aprendizaje llevado a cabo en las aulas. Estas características y algunas posibles estrategias para prevenirlas serían las siguientes:
- Fuerte necesidad de comida: establecer horarios claros en los que puede comer y disminuir su posible acceso a la comida. Concienciar a sus compañeros y a los demás profesionales de sus necesidades alimenticias.
- Presencia de deficiencias o dificultades de aprendizaje: tener en cuenta para adaptar las actividades según sus necesidades.
- Falta de atención: alternar tareas de alto y bajo nivel de actividad, y utilización de materiales motivantes.
- Apnea del sueño: debido a los problemas que puede provocar el cansancio en estos alumnos, a veces, es mejor facilitarles un tiempo para dormir una pequeña siesta, especialmente después del horario establecido para comer.
- Problemas para adaptarse a los cambios: anticipar las rutinas y acontecimientos diarios.
- Aparición de problemas conductuales: crear una pauta de actuación para cada una de las conductas disruptivas que detectemos e informar de ella a todos los profesionales que traten con el niño. El time out es bastante eficaz.
- Inestabilidad emocional y dificultad para expresar sus emociones: evitar lo máximo posible situaciones frustrantes y razonar con ellos, una vez pasada la rabieta, para que hablen sobre lo que sienten
- Conductas obsesivas y perseverancia: especial atención a algunas conductas como rascarse y pellizcarse heridas, e ignorar las preguntas repetitivas.
- Disfunciones en la temperatura corporal y umbral de dolor alto: esto es muy importante porque además está limitado por los problemas en la expresión de las emociones y puede ser el causante de problemas conductuales. Por tanto, es importante la observación.
IntervenciónEn cuanto a la intervención en general a lo largo del ciclo académico del alumno, hay varios puntos que son fundamentales:
- Todos los profesionales deben estar advertidos de las necesidades que presentan estos alumnos.
- Los compañeros deben ser concientes de algunos aspectos, especialmente de la necesidad de que sigan una dieta especial.
- Debemos tener en cuenta sus puntos fuertes para crear a partir de ellos un sistema de refuerzos y para motivarles. En ningún caso se utilizará la comida como recompensa.
- El trabajo de las habilidades sociales es necesario y tiene que estar supervisado para poder integrarse en la vida de la comunidad de forma correcta.
- No tener comida en clase.
- Consulta periódica con la familia.
- Realización de actividades de educación física regularmente.
- Actividades para rehabilitar la motricidad gruesa.
- Trabajar la autoestima.
- Realizar actividades en las que se incentiven las relaciones sociales, el compañerismo y el trabajo en equipo.
Referencias
- ↑ A. Prader, A. Labhart, H. Willi, G. Fanconi:
Ein Syndrom von Adipositas, Kleinwuchs, Kryptorchismus und Idiotie bei Kindern und Erwachsenen, die als Neugeborene ein myotonieartiges Bild geboten haben.
VIII International Congress of Paediatrics, Copenhagen, 1956 - ↑ http://www.ncbi.nlm.nih.gov/books/NBK1330/
- ↑ http://www.ncbi.nlm.nih.gov/books/NBK1330/#pws.Resources
- ↑ Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. 1989 Nov 16;342(6247):281-5
- ↑ Maria J. Mascari, Ph.D., Wayne Gottlieb, M.S., Peter K. Rogan, Ph.D., Merlin G. Butler, M.D., Ph.D., David A. Waller, M.D., John A.L. Armour, Ph.D., Alec J. Jeffreys, Ph.D., Roger L. Ladda, M.D., and Robert D. Nicholls, D.Phil. The Frequency of Uniparental Disomy in Prader-Willi Syndrome — Implications for Molecular Diagnosis. N Engl J Med 1992; 326:1599-1607June 11, 1992
- ↑ Procter M, Chou LS, Tang W, Jama M, Mao R (2006) Molecular diagnosis of Prader-Willi and Angelman Syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin Chem 52:1276–1283.
- ↑ Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR, Whitman BY, Greenberg F. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91:398–402.
- ↑ Gunay-Aygun M, Schwartz S, Heeger S, O'Riordan MA, Cassidy SB. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics. 2001;108:E92.
Bibliografía
- Thompson, Margaret (1996). Thompson & Thompson. Genética en Medicina. Barcelona: Masson. OCLC 44788618.
- Albert García, Marta (1999). El Síndrome de Prade Willi: Guía para familias y profesionales. Instituto de migraciones y servicions sociales, Madrid.
Véase también
 Wikimedia Commons alberga contenido multimedia sobre Síndrome de Prader-Willi. Commons
Wikimedia Commons alberga contenido multimedia sobre Síndrome de Prader-Willi. Commons- Aberración cromosómica
- Deleción
- Medicina genómica
- Síndrome de Angelman
Enlaces externos
- Asociación Síndrome Prader-Willi de Andalucía (ASPWA)
- Asociación Catalana para el Síndrome de Prader-Willi (ACSPW)
- Asociación Española para el Síndrome de Prader-Willi (AESPW)
- Asociación Madrileña para el Síndrome de Prader-Willi (AMSPW)
- Fundación Síndrome de Prader-Willi (FSPW)
- Organización internacional para el SPW (en varios idiomas)
- Otras asociaciones para el SPW de todo el mundo
- Foro Prader-Willi
- Síndrome Prader-Willi en Genetests.org (base de datos de test diagnósticos genéticos)
- Canal PraderWilliVirtual
- Fundación SPINE Argentina
- Diagnóstico temprano
- Alerta Médica, Síndrome de Prader Willi
Categorías:- Educación especial
- Síndromes
- Enfermedades genéticas
- Enfermedades hereditarias
- Enfermedades masculinas
- Enfermedades raras
- Enfermedades epónimas
- Anomalías estructurales cromosómicas

Wikimedia foundation. 2010.