- Aldolasa
-
Aldolasa A[1] 
Tetrámero de aldosa A humanaHUGO 414 Símbolo ALDOA Símbolos alt. ALDA; MGC10942; MGC17716; MGC17767 Datos genéticos Código de gen ALDOA Tipo de gen Gen Codificante Locus Cr. 16 p11.2 Estructura/Función proteica Tamaño 364 (aminoácidos) Productos alternativos 3 isoformas, diferenciadas por su zona 5'-UTR Bases de datos Números de accesión X05236 Número EC 4.1.2.13 Entrez 226 OMIM 103850 RefSeq NM_000034 UniProt P04075 Aldolasa B[2] 
HUGO 417 Símbolo ALDOB Datos genéticos Código de gen ALDOB Tipo de gen Gen codificante Locus Cr. 9 q21.3-q22.2 Estructura/Función proteica Tamaño 364 (aminoácidos) Bases de datos Número EC 4.1.2.13 Entrez 229 OMIM 612724 RefSeq NM_000035 UniProt P05062 Aldolasa C[3] 
HUGO 418 Símbolo ALDOC Datos genéticos Código de gen ALDOC Tipo de gen Gen codificante Locus Cr. 17 [1] Estructura/Función proteica Tamaño 364 (aminoácidos) Bases de datos Número EC 4.1.2.13 Entrez 230 OMIM 103870 RefSeq NM_005165 UniProt P09972 La aldolasa (EC 4.1.2.13) es una enzima que participa en la glucólisis. La aldolasa cataliza la escisión de la fructosa-1,6-bisfosfato en dos triosas, dihidroxiacetona fosfato y gliceraldehído 3-fosfato.[4]
- D-fructosa-1,6-bisfosfato
 dihidroxiacetona fosfato + D-gliceraldehído-3-fosfato
dihidroxiacetona fosfato + D-gliceraldehído-3-fosfato
También cataliza la rotura reversible de la fructosa-1-fosfato en gliceraldehído y dihidroxiacetona fosfato.
- D-fructosa-1-fosfato dihidroxiacetona fosfato + D-gliceraldehído
En el ser humano se conocen tres isozimas de la aldolasa (aldolasa A, aldolasa B y aldolasa C) codificadas por tres genes diferentes y que se expresan de forma diferente durante el desarrollo. Pequeñas diferencias en la estructura de las isozimas resultan en diferentes actividades sobre los dos sustratos: fructosa-1,6-bisfosfato y fructosa-1-fosfato. La aldolasa B no exhibe preferencia sobre los sustratos y, por tanto, cataliza ambas reacciones. En cambio las aldolasas A y C prefieren la fructosa-1,6-bisfosfato.[5]
Contenido
Mecanismo
En la aldolasa mamífera, los residuos catalíticos clave que participan en la reacción son lisina y tirosina. La tirosina actúa como un aceptor eficiente de hidrógeno mientras que la lisina se une covalentemente y estabiliza el intermedio. Muchas bacterias usan dos iones magnesio en vez de la lisina.
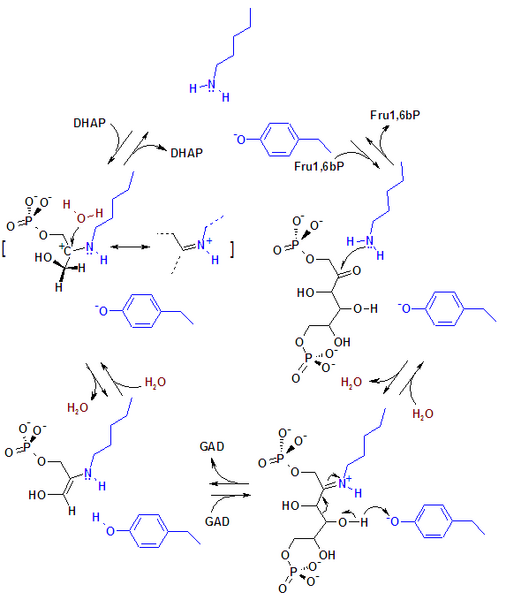
Mecanismos de reacción de la aldolasa. 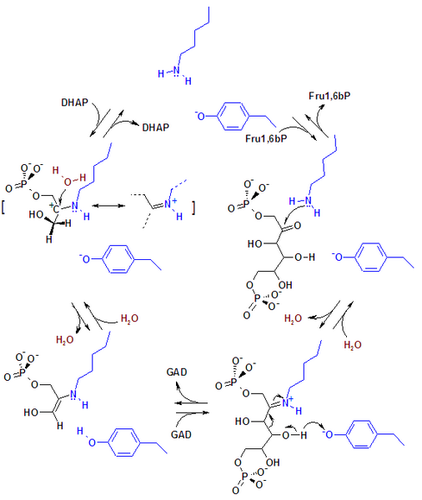 Figura 1. Mecanismo de reacción de la aldolasa para la rotura de la fructosa-1,6-bisfosfato. Las cadenas laterales de los residuos del sitio activo están mostradas en azul.
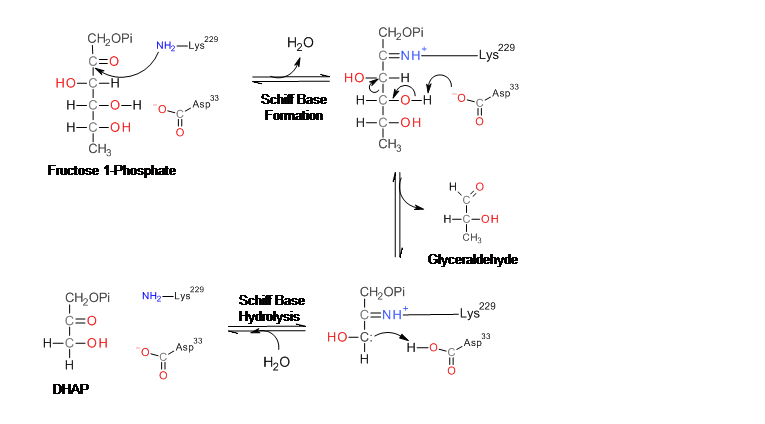
Figura 1. Mecanismo de reacción de la aldolasa para la rotura de la fructosa-1,6-bisfosfato. Las cadenas laterales de los residuos del sitio activo están mostradas en azul.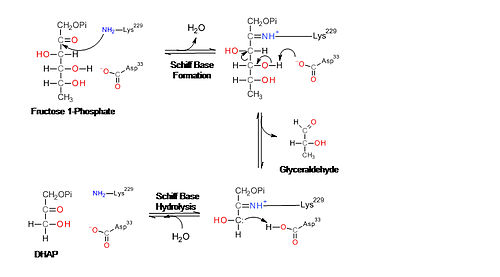 Figura 2. Mecanismo de la aldolasa para la rotura de la fructosa-1-fosfato para producir dihidroxiacetona fosfato y gliceraldehído.
Figura 2. Mecanismo de la aldolasa para la rotura de la fructosa-1-fosfato para producir dihidroxiacetona fosfato y gliceraldehído.La enzima fructosa bisfosfato aldolasa rompe una fructosa de 6 átomos de carbono en dos productos de 3 átomos de carbono en una reacción aldólica reversible. Esta reacción está caracterizada por la formación de una base Schiff intermedia con un residuo lisina del sitio activo de la enzima. La formación de la base Schiff es el hecho diferenciador entre la enzima clase I producida por los animales y la enzima clase II producida por los hongos y bacterias. Después de la formación de la base Schiff, el cuarto grupo hidroxilo del esqueleto fructosa es deprotonado por un residuo aspartato que resulta en la rotura del aldol. La hidrólisis de la base Schiff produce dos productos de 3 átomos de carbono.[6]
La ΔG°’ de esta reacción es +23.9 kJ/mol. A pesar de que parezca que la energía libre sea demasiado elevada para que la reacción ocurra, se ha encontrado que bajo condiciones fisiológicas, la ΔG de la reacción está cerca e incluso por debajo de cero. Por ejemplo, la ΔG de esta reacción bajo condiciones fisiológicas en los eritrocitos es de -0.23 kJ/mol.[6]
Aldolasa A
La aldolasa A se encuentra en el embrión en desarrollo y se produce en grandes cantidades en el músculo adulto. Se presenta como un homotetrámero.[7] La expresión de la aldolasa A es reprimida en el hígado adulto, riñones e intestino. Se encuentra en niveles similares a la aldolasa C en el cerebro y en otros tejidos del sistema nervioso. El splicing alternativo del gen de la aldolasa A resulta en múltiples variantes que codifican la misma proteína.[8] Se ha demostrado que la aldolasa A interacciona con PLD2,[9] , SNX9 y WAS.[7]
Los defectos en aldolasa A son causa de la enfermedad de almacenamiento del glucógeno tipo 12 (GSD12), conocida también como deficiencia de la aldolasa de los glóbulos rojos. Es un desorden metabólico asociado con un incremento en glucógeno hepático y anemia hemolítica. Puede producir miopatía con intolerancia al ejercicio y rabdomiolisis.[7]
Aldolasa B
En los mamíferos, la aldolasa B se expresa preferentemente en el hígado. En los humanos, la aldolasa B es codificada por el gen ALDOB localizado en el cromosoma 9. Este gen tiene 14,500 pares de bases y contiene 9 exones.[10] [11] [12] Los defectos en este gen se han identificado como causa de la intolerancia hereditaria a la fructosa.[13]
Estructura
La aldolasa B es una enzima homotetramérica compuesta de cuatro subunidades de peso molecular 36 kDa con simetría 222 local. Cada subunidad contiene un barril α/β de 8 filamentos que contiene la Lys-229 que es el residuo que forma la base Schiff necesaria para la catálisis.[14] [15]
A pesar de que la mayoría de la estructura de la enzima aldolasa está conservada en las tres isozimas, se han identificado cuatro regiones que son altamente variables entre las tres isozimas. A estas regiones se les ha denominado como regiones específicas de la isozimas (ISR1-4). Se cree que estas regiones proporcionan a las isozimas sus especificidades y diferencias estructurales. Las ISRs 1-3 se encuentran todas en el exon 3 del gen de la aldolasa B. La ISR 4 es la más variable de las cuatro y se encuentra en la región C-terminal final de la proteína.[5]
Las ISRs 1-3 se encuentran predominantemente en la superficie de la enzima y no coinciden con el sitio activo, indicando que las ISRs pueden cambiar la especificidad por el sustrato o causar las interacciones de la región C-terminal con el sitio activo.[15] Una teoría reciente sugiere que las ISRs pueden permitir diferentes dinámicas conformacionales de la enzima aldolasa que influyen en su especificidad.[16]
Fisiología
La aldolasa B juega un papel crucial en el metabolismo de los carbohidratos ya que cataliza una de las principales etapas de la glucólisis y gluconeogénesis. También juega un papel importante en el metabolismo de la fructosa que se produce principalmente en el hígado, corteza adrenal y mucosa del intestino delgado. Cuando la fructosa es absorbida, es fosforilada por la fructokinasa para formar fructosa-1-fosfato. La aldolasa B entonces cataliza la rotura de la fructosa-1-fosfato en gliceraldehído y dihidroxiacetona fosfato. El gliceraldehído es fosforilado por la triosa kinasa para formar gliceraldehído-3-fosfato. Tanto la dihidroxiacetona como el gliceraldehído-3-fosfato pueden ser usados en la glicólisis y gluconeogénesis, es decir, pueden ser modificadas para convertirse en glucosa o en piruvato.[17]
A pesar de que el mecanismo de regulación de la aldolasa B es desconocido, se ha encontrado que un incremento en los carbohidratos proveniente de la dieta y un decremento en la concentración de glucagón provocan un incremento en la transcripción del gen ALDOB.[18] [19]
Patología
Las mutaciones genéticas que provocan defectos en aldolasa B resultan en una condición llamada intolerancia hereditaria a la fructosa (HFI). Debido a la pérdida de aldolasa B funcional, los organismos con HFI no pueden procesar adecuadamente fructosa-1-fosfato produciendo la acumulación de este metabolito en los tejidos del cuerpo. Además de ser tóxico para los tejidos celulares, altos niveles en fructosa-1-fosfato atrapan el fosfato en una forma no usable y, por tanto, no es devuelto a las reservas de fosfato resultando en una reducción en la concentración de fosfato y ATP. La falta de fosfato disponible causa el cese de la glucogenólisis en el hígado que resulta en hipoglicemia.[20] La acumulación de fructosa-1-fosfato también inhibe la gluconeogénesis, reduciendo adicionalmente la cantidad de glucosa disponible. La pérdida de ATP produce una multitud de problemas incluyendo la inhibición de la síntesis de proteínas y disfunción hepática y renal. Sin embargo, la prognosis del paciente es buena en casos de HFI. Evitando alimentos que contengan fructosa, sucrosa y sorbitol, los pacientes pueden vivir sin síntomas.[17]
La HFI es un desorden autosomal recesivo hereditario. Se han identificado aproximadamente 30 mutaciones que causan HFI, y estas mutaciones combinadas resultan en una frecuencia de la HFI de 1 enfermo cada 20.000 nacimientos.[17] [21] Los alelos mutantes son resultado de un número de diferentes tipos de mutaciones incluyendo sustituciones de pares de bases y pequeños borrados. La mutación más común es la A149P que es una transversión de guanina a citosina en el exón 5, resultando en la sustitución de la alanina a prolina en la posición 149. Este alelo específico mutante comprende el 53% de los alelos de la HFI.[22] Otras mutaciones que resultan en HFI son menos frecuentes y a veces correlacionadas con orígenes ancestrales.[23]
Aldolasa C
Se expresa específicamente en el hipocampo y en las células de Purkinje del cerebro. Se presenta como un homotetrámero. Interacciona con ATP6V1E1. Puede que interaccione con PLD2.[24]
Referencias
- ↑ «ALDOA». Consultado el 10 de noviembre de 2011.
- ↑ «ALDOB». Consultado el 10 de noviembre de 2011.
- ↑ «ALDOC». Consultado el 10 de noviembre de 2011.
- ↑ «ENZYME entry: EC 4.1.2.13». Consultado el 7 de noviembre de 2011.
- ↑ a b Dalby AR, Tolan DR, Littlechild JA (November 2001). «The structure of human liver fructose-1,6-bisphosphate aldolase». Acta Crystallogr. D Biol. Crystallogr. 57 (Pt 11): pp. 1526–33. doi:. PMID 11679716.
- ↑ a b Garrett RH and Grisham CM (2010). Biochemistry 4th Edition. Brooks/Cole.
- ↑ a b c «Fructose-bisphosphate aldolase A». Consultado el 10 de noviembre de 2011.
- ↑ «Entrez Gene: ALDOA aldolase A, fructose-bisphosphate».
- ↑ Kim, Jong Hyun; Lee Sukmook, Kim Jung Hwan, Lee Taehoon G, Hirata Masato, Suh Pann-Ghill, Ryu Sung Ho (Mar. 2002). «Phospholipase D2 directly interacts with aldolase via Its PH domain». Biochemistry (United States) 41 (10): pp. 3414–21. doi:. ISSN 0006-2960. PMID 11876650.
- ↑ «Entrez Gene: ALDOB aldolase B, fructose-bisphosphate».
- ↑ Henry I, Gallano P, Besmond C, Weil D, Mattei MG, Turleau C, Boué J, Kahn A, Junien C (July 1985). «The structural gene for aldolase B (ALDB) maps to 9q13----32». Ann. Hum. Genet. 49 (Pt 3): pp. 173–80. doi:. PMID 3000275.
- ↑ Tolan DR, Penhoet EE (June 1986). «Characterization of the human aldolase B gene». Mol. Biol. Med. 3 (3): pp. 245–64. PMID 3016456.
- ↑ Cox TM (January 1994). «Aldolase B and fructose intolerance». FASEB J. 8 (1): pp. 62–71. PMID 8299892.
- ↑ Sygusch J, Beaudry D, Allaire M (November 1987). «Molecular architecture of rabbit skeletal muscle aldolase at 2.7-A resolution». Proc. Natl. Acad. Sci. U.S.A. 84 (22): pp. 7846–50. doi:. PMID 3479768.
- ↑ a b Pezza JA, Choi KH, Berardini TZ, Beernink PT, Allen KN, Tolan DR (May 2003). «Spatial clustering of isozyme-specific residues reveals unlikely determinants of isozyme specificity in fructose-1,6-bisphosphate aldolase». J. Biol. Chem. 278 (19): pp. 17307–13. doi:. PMID 12611890.
- ↑ Pezza JA, Stopa JD, Brunyak EM, Allen KN, Tolan DR (November 2007). «Thermodynamic Analysis Shows Conformational Coupling/Dynamics Confers Substrate Specificity in Fructose-1,6-bisphosphate Aldolase». Biochemistry 46 (45): pp. 13010–8. doi:. PMID 17935305.
- ↑ a b c Inborn Metabolic Diseases, Fourth Revised Edition. Springer Berlin Heidelberg. 2006.
- ↑ Gomez PF, Ito K, Huang Y, Otsu K, Kuzumaki T, and Ishikawa K (November 1994). «Dietary and hormonal regulation of aldolase B gene transcription in rat liver». Arch Biochem Biophys 314 (2): pp. 307–14. doi:. PMID 7979370.
- ↑ Munnich A, Besmond C, Darquy S, et al. (March 1985). «Dietary and hormonal regulation of aldolase B gene expression». J. Clin. Invest. 75 (3): pp. 1045–52. doi:. PMID 2984252.
- ↑ Bouteldja N, Timson DJ (April 2010). «The biochemical basis of hereditary fructose intolerance». J. Inherit. Metab. Dis. 33 (2): pp. 105–12. doi:. PMID 20162364.
- ↑ Esposito G, Vitagliano L, Santamaria R, Viola A, Zagari A, Salvatore F (November 2002). «Structural and functional analysis of aldolase B mutants related to hereditary fructose intolerance». FEBS Lett. 531 (2): pp. 152–6. doi:. PMID 12417303.
- ↑ Malay AD, Allen KN, and Tolan DR (March 2005). «Structure of the thermolabile mutant aldolase B, A149P: molecular basis of hereditary fructose intolerance». J Mol Biol. 347 (1): pp. 135–44. doi:. PMID 15733923.
- ↑ Tolan DR (1995). «Molecular basis of hereditary fructose intolerance: mutations and polymorphisms in the human aldolase B gene». Hum. Mutat. 6 (3): pp. 210–8. doi:. PMID 8535439.
- ↑ «Fructose-bisphosphate aldolase C». Consultado el 10 de noviembre de 2011.
Categorías:- Genes del cromosoma 16
- Genes del cromosoma 9
- Genes del cromosoma 17
- Liasas
- Glucólisis
- Proteínas humanas
- D-fructosa-1,6-bisfosfato
Wikimedia foundation. 2010.