- Reacción aldólica
-
La reacción aldólica es una reacción química de formación de enlaces carbono-carbono en química orgánica.[1] [2] [3] En su forma normal, la reacción aldólica involucra la adición nucleofílica del enolato de una cetona a un aldehído, para formar una β-hidroxicetona, o "aldol" (aldehído + alcohol), una unidad estructural que se encuentra en muchas moléculas presentes en moléculas de origen natural y en fármacos.[4] [5] [6] Algunas veces, el producto de la adición aldólica pierde una molécula de agua durante la reacción para formar una cetona α,β-insaturada, lo que se conoce como condensación aldólica. La reacción aldólica fue descubierta independientemente por Charles-Adolphe Wurtz[7] [8] [9] y Alexander Porfyrevich Borodin en 1872.[10] Borodin observó la dimerización aldólica a 3-hidroxibutanal a partir de acetaldehído bajo condiciones ácidas. La reacción aldólica se usa ampliamente en la producción a gran escala de productos químicos que servirán en sucesivos procesos como materias primas, tales como el pentaeritritol[11] y en la industria farmacéutica para la síntesis de medicamentos de pureza óptica. Por ejemplo, la ruta que inicialmente empleó la empresa Pfizer para sintetizar el fármaco anticolesterolémico Lipitor (atorvastatina), aprobado en 1996, empleaba dos reacciones aldólicas, permitiendo la producción de cantidades del fármaco en la escala de los multigramos.[12] [13]
-
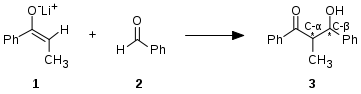 Adición aldólica típica: un enolato de cetona típico 1, actuando como nucleófilo, se adiciona al carbono electrofílico de un aldehído 2, formándose un nuevo enlace carbono-carbono, obteniéndose el producto aldólico 3.
Adición aldólica típica: un enolato de cetona típico 1, actuando como nucleófilo, se adiciona al carbono electrofílico de un aldehído 2, formándose un nuevo enlace carbono-carbono, obteniéndose el producto aldólico 3. -
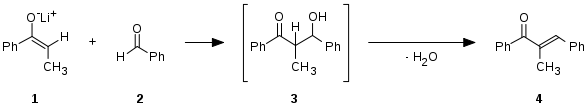 Condensación aldólica típica: un enolato de cetona 1, se adiciona al carbono electrofílico de un aldehído 2, formándose el producto de adición 3. Este producto intermedio pierde una molécula de agua, obteniéndose el producto de condensación 4.
Condensación aldólica típica: un enolato de cetona 1, se adiciona al carbono electrofílico de un aldehído 2, formándose el producto de adición 3. Este producto intermedio pierde una molécula de agua, obteniéndose el producto de condensación 4.
La unidad estructural aldólica es especialmente común en los policétidos, una familia de productos naturales de los cuales derivan muchos fármacos, incluyendo el potente inmunosupresor FK506, los antibióticos tetraciclinas, y el agente antifúngico anfotericina B. La amplia investigación en la reacción aldólica ha producido métodos altamente eficientes que lo permiten, ya que de otro modo sería muy complicada síntesis total de muchos policétidos en el laboratorio.[14] Esto es importante porque muchos policétidos, junto con otras moléculas biológicamente activas, están presentes en la naturaleza en cantidades no prácticas y pequeñas para investigaciones posteriores. La síntesis de muchos compuestos, otrora considerada casi imposible, puede ser efectuada casi rutinariamente a escala de laboratorio, y se acerca a la viabilidad económica a gran escala en algunos casos, tales como el agente antitumoral altamente activo discodermolida. En bioquímica, la reacción aldólica es uno de los pasos clave de la glicólisis, donde es catalizada por enzimas denominadas aldolasas.
La reacción aldólica es particularmente valiosa en síntesis orgánica porque conduce a la formación de productos con dos nuevos centros estereogénicos (en el carbono-α y en el carbono-β del aducto aldólico, marcado con asteriscos en el esquema anterior). Los métodos modernos, descritos más adelante, permiten controlar la configuración relativa y absoluta de estos centros. Esto es de particular importancia cuando se sintetizan fármacos, dado que las moléculas con la misma conectividad estructural pero diferente estereoquímica suelen tener propiedades biológicas ampliamente distintas.
Se puede emplear una variedad de nucleófilos en la reacción aldólica, incluyendo el enol, enolato, éter de enol de las cetonas, aldehídos y muchos otros compuestos carbonílicos. Generalmente, el compañero electrofílico es un aldehído, aunque existen muchas variaciones, como la reacción de Mannich. Cuando el nucleófilo y el electrófilo son distintos (el caso más frecuente), la reacción es denominada reacción aldólica cruzada (en oposición a la que forma dímeros en una dimerización aldólica).
 Puesta a punto experimental típica para una reacción aldólica. Una solución de diisopropilamida de litio (LDA) en tetrahidrofurano (THF) (en el matraz de la derecha) se agrega lentamente a una solución de propionato de tert-butilo en el matraz de la izquierda, formando su enolato de litio. Luego puede agregarse un aldehído para iniciar la reacción de adición aldólica. Ambos matraces están sumergidos en un baño refrigerante de hielo seco/acetona (-78 °C), cuya temperatura está siendo monitorizada por un termopar (el cable de la izquierda).
Puesta a punto experimental típica para una reacción aldólica. Una solución de diisopropilamida de litio (LDA) en tetrahidrofurano (THF) (en el matraz de la derecha) se agrega lentamente a una solución de propionato de tert-butilo en el matraz de la izquierda, formando su enolato de litio. Luego puede agregarse un aldehído para iniciar la reacción de adición aldólica. Ambos matraces están sumergidos en un baño refrigerante de hielo seco/acetona (-78 °C), cuya temperatura está siendo monitorizada por un termopar (el cable de la izquierda).
Contenido
Mecanismos
La reacción aldólica puede proceder mediante dos mecanismos fundamentalmente distintos. En el "mecanismo enólico", los compuestos carbonílicos, tales como los aldehídos y cetonas, pueden ser convertidos a enoles o éteres de enol. Estos compuestos, al ser nucleofílicos en el carbono-α, pueden atacar a los compuestos carbonílicos protonados especialmente reactivos, tales como los aldehídos protonados. En el "mecanismo del enolato", los compuestos carbonílicos, al tener átomos de hidrógeno ácidos, pueden ser deprotonados para formar enolatos, que son mucho más nucleofílicos que los enoles o los éteres de enol y pueden atacar a los electrófilos directamente. El electrófilo usual es un aldehído, dado que las cetonas son mucho menos reactivas.
Si las condiciones son particularmente fuertes (por ejemplo, metóxido de sodio, hidróxido de sodio, reflujo), puede producirse la condensación, pero esto generalmente puede evitarse con reactivos que requieren condiciones moderadas o bajas temperaturas (por ejemplo, LDA -una base fuerte-, THF, -78 °C). Aunque la adición aldólica generalmente avanza hasta casi completarse, la reacción no es irreversible, pues el tratamiento de los aductos aldólicos con bases fuertes generalmente induce la ruptura retro-aldólica (produciendo los materiales iniciales). Las condensaciones aldólicas son irreversibles.
Mecanismo enólico
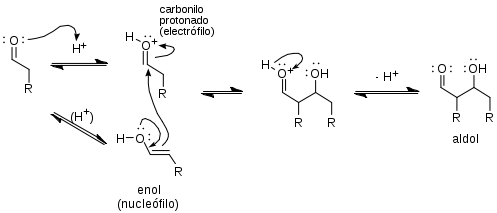
Cuando se usa un catalizador ácido, la etapa inicial en el mecanismo de reacción involucra la tautomerización del compuesto carbonílico para formar el enol. El ácido también sirve para activar el grupo carbonílico de otra molécula por protonación, haciéndolo altamente electrofílico. El enol es nucleofílico en el carbono-α, permitiéndole atacar el compuesto carbonílico protonado, conduciendo al aldol después de la deprotonación. Éste suele deshidratarse para producir el compuesto carbonílico insaturado. El esquema muestra una autocondensación catalizada por ácido típica de un aldehído.
- Mecanismo aldólico catalizado por ácido
- Deshidratación catalizada por ácido
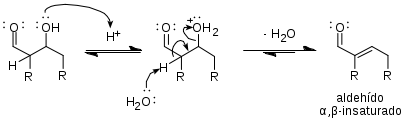
Mecanismo del enolato
Si el catalizador es una base moderada como el ion hidróxido o un alcóxido, la reacción aldólica procede vía el ataque nucleofílico del enolato estabilizado por resonancia al grupo carbonilo de otra molécula. El producto es la sal de alcóxido del producto aldólico. A continuación se forma el aldol mismo, y puede sufrir deshidratación para producir el compuesto carbonílico insaturado. El esquema muestra un mecanismo simple para la reacción aldólica catalizada por bases de un aldehído consigo mismo.
Reacción aldólica catalizada por bases (mostrada usando −OCH3 como base)
Deshidratación catalizada por bases (algunas veces escrita en un solo paso)
Aunque sólo se requiere una cantidad catalítica de base en algunos casos, el procedimiento más usual es usar una cantidad estequiométrica de base fuerte tal como el LDA o NaHMDS. En este caso, la formación de enolato es irreversible, y el producto aldólico no se forma hasta que el alcóxido metálico del producto aldólico es protonado en un paso posterior.
Modelo de Zimmerman-Traxler
Se conocen formas más refinadas del mecanismo. En 1957, Zimmerman y Traxler propusieron que algunas reacciones aldólicas tienen estados de transición de seis miembros en conformación de silla.[15] Esto es conocido como modelo de Zimmerman-Traxler. Los E-enolatos conducen a la formación de productos anti, mientras que los Z-enolatos conducen a la formación de productos syn. Los factores que controlan la selectividad son la preferencia para colocar los sustituyentes ecuatorialmente en los estados de transición de seis miembros y evitar interacciones syn-pentano respectivamente.[16] E y Z se refieren a las relaciones estereoquímicas cis-trans entre los átomos de oxígeno de enolato que tienen el contraión positivo, y el grupo de máxima prioridad en el carbono α. En realidad, sólo algunas especies como el litio y el boro siguen el modelo de Zimmerman-Traxler. En consecuencia, en algunos casos el resultado estereoquímico de la reacción puede ser impredecible.
Control en la reacción aldólica
El problema
El problema del "control" en la adición aldólica es demostrado mejor por un ejemplo. Considérese el resultado de esta reacción hipotética:
En esta reacción, dos cetonas asimétricas se condensan usando etóxido de sodio. La basicidad del etóxido de sodio es tal que no puede deprotonar completamente ninguna de las cetonas, pero puede producir pequeñas cantidades de enolato de sodio para ambas. esto significa que, además de ser potenciales electrófilos aldólicos, ambas cetonas pueden actuar también como nucleófilos por medio de su enolato de sodio. Dos electrófilos y dos nucleófilos pueden conducir potencialmente a cuatro productos posibles:
En consecuencia, si se desea obtener sólo uno de los productos cruzados, la adición aldólica debe ser "controlada".
Acidez
Si uno de los reactivos es considerablemente más ácido que el otro, el control puede ser automático. El protón más ácido es abstraído por la base y se forma un enolato. Este tipo de control sólo sirve si la diferencia de acidez es suficientemente grande y no se usa un exceso de base en la reacción. El control más simple se puede dar si sólo uno de los reactivos tiene protones ácidos y sólo esta molécula forma el enolato. Por ejemplo, la adición de malonato de dietilo al benzaldehído sólo conduciría a la formación de un producto:
En este caso, los protones metilénicos doblemente activados del malonato serán preferentemente deprotonados por el etóxido de sodio, y formarán cuantitativamente el enolato de sodio. Dado que el benzaldehído no tiene átomos de hidrógeno en posición α, sólo es posible una combinación nucleófilo-electrófilo; entonces se ha conseguido el control. Esta aproximación combina dos elementos del control: acidez aumentada de los átomos de hidrógeno α en el nucleófilo, y la falta de átomos de hidrógeno α en el electrófilo.
Orden de adición
Una solución común es la formación del enolato de uno de los reactivos primero, y luego agregar el otro reactivo bajo control cinético de la reacción.[17] El control cinético significa que la reacción directa de adición aldólica debe ser significativamente más rápida que la reacción inversa retro-aldólica. Para que esta aproximación sea exitosa, también deben satisfacerse otras dos condiciones: debe ser posible la formación cuantitativa del enolato del primer reactivo, y la reacción directa aldólica debe ser significativamente más rápida que la transferencia de la forma de enolato de un reactivo al otro. Algunas condiciones de control cinético comunes implican la formación del enolato de una cetona con LDA a -78 °C, seguido de la lenta adición de un aldehído.
Enolatos
Formación
El enolato puede ser formando usando una base fuerte ("condiciones duras") o un ácido de Lewis y una base débil ("condiciones suaves"). Para que suceda la deprotonación, el requerimiento estereoelectrónico es que el enlace sigma Cα-H sea capaz de traslaparse con el orbital π* del grupo carbonilo.
Geometría
Se han desarrollado amplios estudios sobre la formación de enolatos bajo diferentes condiciones. Es posible generar, en la mayoría de casos, le geometría del enolato deseada:[18]
Para las cetonas, la mayoría de condiciones de enolización produce los Z enolatos. Para los ésteres, la mayoría de condiciones de enolización produce los E enolatos. La adición de HMPA invierte la estereoselectividad de la deprotonación.
La formación estereoselectiva de los enolatos ha sido racionalizada mediante el denominado modelo Irlanda,[19] [20] [21] [22] aunque su validez es algo cuestionable. En muchos casos, no se conoce qué intermediarios son monoméricos o oligoméricos en naturaleza; a pesar de ello, el modelo Irlanda sigue siendo una herramienta útil para entender los enolatos.
En el modelo Irlanda, se asume que la deprotonación procede por un estado de transición monomérico de seis miembros. Los dos sustituyentes más grandes en el electrófilo adoptan una disposición ecuatorial en el estado de transición favorecida, conduciendo a una preferencia por los E enolatos. El modelo falla claramente en muchos casos; por ejemplo, si la mezcla solvente es cambiada de THF a 23% HMPA-THF la geometría del enolato se invierte inexplicablemente.
Enolato cinético vs enolato termodinámico
Si una cetona asimétrica es atacada por una base, puede formar dos enolatos regioisoméricos (ignorando la geometría del enolato). Por ejemplo:
Se considera al enolato trisustituido como el enolato cinético, mientras que el enolato tetrasustituido es considerado como el enolato termodinámico. El átomo de carbono α deprotonado para formar el enolato cinético está menos protegido, y en consecuencia se deprotona más fácilmente. En general, las olefinas tetrasustituidas son más estables que las olefinas trisustituidas debido a la estabilización hiperconjugativa. La proporción de regioisómeros del enolato está altamente influenciada por la base seleccionada. Para el ejemplo anterior, el control cinético debe establecerse con LDA a -78 °C, con un resultado de una selectividad cinética de 99:1, mientras que en el caso del enolato termodinámico, el control termodinámico debe ser establecido con trifenilmetillitio a temperatura ambiente, dando una selectividad de 90:10.
En general, los enolatos cinéticos son favorecidos por temperaturas frías, enlaces metal-oxígeno con mayor carácter iónico y deprotonación rápida usando un ligero exceso de una base fuerte y con gran impedimento estérico; los enolatos termodinámicos se ven favorecidos por temperaturas altas, enlaces metal-oxígeno con mayor carácter covalente, y mayor tiempo para que alcance el equilibrio utilizando una cantidad ligeramente sub-estequiométrica de base fuerte. El uso de una cantidad sub-estequiométrica de base permite que una pequeña fracción de compuesto carbonílico, no enolizado, equilibre al enolato hacia el regioisómero termodinámico, actuando como un transportador de protones.
Estereoselectividad
La reacción aldólica es particularmente útil porque se generan dos nuevos centros estereogénicos en una reacción. La extensa investigación efectuada ha permitido entender el mecanismo de reacción y mejorar la selectividad observada bajo diferentes condiciones. La convención syn/anti es usada comúnmente para denotar la estereoquímica relativa en los átomos de carbono α y β.
La convención se aplica cuando se agregan nucleófilos de propionato (u orden mayor) a aldehídos. El grupo R de la cetona y el grupo R del aldehído se alínean en zigzag en el plano del papel, y la disposición es syn o anti, dependiendo de que si están al mismo lado o a lados opuestos de la cadena principal.
Enolatos E vs. Z
No hay diferencia significativa entre el nivel de estereoinducción observada con los enolatos E y Z:[18]
Ion metálico
El catión metálico del enolato puede desempeñar un importante papel para determinar el nivel de estereoselectividad en la reacción aldólica. Se usa boro frecuentemente porque su longitud de enlace es significativamente menor que la de otros metales como el litio, el aluminio o el magnesio. Por ejemplo, los enlaces boro-carbono y boro-oxígeno tienen longitudes de 1,4-1,5 Å y 1,5–1,6 Å de longitud respectivamente, mientras que las longitudes de enlace típicas metal-carbono y metal-oxígeno se encuentran entre 1,9–2,2 Å y 2,0–2,2 Å respectivamente. Esto tiene efecto en el "fortalecimiento" del estado de transición:[23]
Estereoselectividad: Estereocentro α en el enolato
La reacción aldólica puede exhibir "estereocontrol basado en el sustrato" en el que la quiralidad existente en cualquiera de los reactivos influye en el resultado estereoquímico de la reacción. Esto ha sido estudiado ampliamente, y en muchos casos, se puede predecir el sentido de la inducción asimétrica, y en ocasiones el nivel absoluto de diastereoselectividad. Si el enolato contiene un estereocentro en la posición α, se puede realizar un excelente estereocontrol.
En el caso de un E enolato, el elemento dominante de control es la tensión alílica 1,3, donde, en el caso de un Z enolato, el elemento de control dominante es impedir las interacciones diaxiales 1,3. El modelo general es presentado a continuación:
Para claridad, el estereocentro en el enolato ha sido epimerizado; en realidad, la diastereocara opuesta del aldehído debía haber sido atacada. En ambos casos, el diastereómero syn-1,3 está favorecido. Hay muchos ejemplos de este tipo de estereocontrol:[24]
Estereoselectividad: estereocentro en α en el electrófilo
Cuando los enolatos atacan a un aldehído con un estereocentro en α, es también posible un excelente estereocontrol. La observación general es que los enolatos E exhiben selección diasterofacial Felkin, mientras que los enolatos Z exhiben selectividad anti-Felkin. El modelo general es el siguiente:[25] [26]
Ya que los enolatos Z deben reaccionar a través de un estado de transición que contiene o una interacción desestabilizante syn-pentano o un rotámero anti-Felkin, los enolatos Z exhiben menores niveles de diastereoselectividad en este caso. Algunos ejemplos se presentan debajo:[27] [28]
Estereoselectividad: modelo combinado para la estereoinducción
Si tanto el enolato como el aldehído contienen quiralidad pre-existente, el resultado de la "doble estereodiferenciación" de la reacción aldólica puede ser predicha usando un modelo estereoquímico fusionado, que toma en cuenta las interacciones faciales en el enolato, la geometría del enolato, y las interacciones faciales en el aldehído.[29] Algunos ejemplos de la aplicación de este modelo se dan a continuación:[28]
Química de la oxazolidinona de Evans
La síntesis orgánica moderna requiere de la síntesis de compuestos en forma enantiopura. Dado que la reacción de adición aldólica crea dos nuevos estereocentros, pueden formarse hasta cuatro estereoisómeros.
Se han desarrollado muchos métodos que permiten tanto el control de la estereoquímica relativa (syn o anti) y absoluta (R o S).
Un método ampliamente usado es el método de la oxazolidinona de acilo de Evans.[30] [31] Fue desarrollado a fines de la década de 1970 y principios de 1980 por David A. Evans y sus colaboradores. El método funciona al crear un enolato quiral al unir un auxiliar quiral. La quiralidad pre-existente del auxiliar es transferida al aducto aldólico mediante una reacción aldólica diastereoselectiva. Después de la subsecuente remoción del auxiliar, se obtiene el estereoisómero aldólico deseado.
En el caso del método de Evans, el auxiliar quiral unido es una oxazolidinona, y el compuesto carbonílico resultante es una imida. Hay un buen número de oxazolidinonas fácilmente disponibles en ambas formas enantioméricas. Éstas pueden costar entre los 10 y los 20 dólares estadounidenses por gramo, con lo que son relativamente caras.
La acilación de una oxazolidinona es un procedimiento conveniente, y es referida informalmente como "loading done". Los Z-enolatos, que conducen a aductos aldólicos syn, pueden ser formados usando enolización suave mediada por boro:[32]
Frecuentemente puede obtenerse sólo un diastereómero por una cristalización del aducto aldólico. Los aductos aldólicos anti no pueden ser obtenidos apropiadamente mediante el método de Evans. A pesar del costo y la limitación de conducir solamente a aductos syn, la superior fiabilidad del método, facilidad de uso y versatilidad lo convierten en el método de elección en muchas situaciones. Muchos otros métodos están disponibles para la ruptura del auxiliar:[33]
A partir de la construcción de la imida, pueden realizarse reacciones de adición aldólicas selectivas syn y anti, permitiendo el ensamblaje de tres de los cuatro posibles posiciones estereoselectivas: selectivo syn:[34] y selectivo anti:[35]
En las reacciones syn-selectivas, ambos métodos de enolización producen el enolato Z, como se espera; sin embargo, el resultado estereoquímico de la reacción está controlado por el estereocentro metilo, más que por la quiralidad de la oxazolidinona. Los métodos descritos permiten el ensamblaje estereoselectivo de policétidos.
Química aldólica moderna
La metodología reciente permite llevar a cabo una variedad de reacciones aldólicas más amplia, frecuentemente con una cantidad catalítica de ligando quiral. Cuando las reacciones emplean pequeñas cantidades de ligandos enantioméricamente puros para inducir a la formación de productos igualmente enantioméricamente puros, las reacciones suelen ser denominas "catalíticas, asimétricas"; hay muchas reacciones aldólicas asimétricas catalíticas disponibles.
Reacciones aldólicas acetato
Una limitación clave a la aproximación por auxiliar quiral es la incapacidad de las N-acetil imidas para reaccionar selectivamente. Una aproximación usada inicialmente era emplear un grupo tioéter temporal:[33] [36]
Reacción aldólica de Mukaiyama
La reacción aldólica de Mukaiyama es la adición nucleofílica de éteres enólicos de sililo a un aldehído, catalizada por ácidos de Lewis, tales como el trifluoruro de boro o el cloruro de titanio.[37] [38] La reacción aldólica de Mukaiyama no sigue el modelo de Zimmerman-Traxler. Carreira describió una metodología asimétrica particularmente útil con acetales de sililcetena debido a su elevado grado de enantioselectividad y sustratos susceptibles.[39]
El método funciona con aldehídos alifáticos no ramificados, que suelen ser electrófilos pobres para los procesos asimétricos, catalíticos. Esto puede deberse a la pobre diferenciación electrónica y estérica entre sus enantiocaras.
El proceso aldólico vinílico análogo de Mukaiyama puede ser efectuado en forma catalítica y asimétrica. El ejemplo mostrado a continuación funciona eficientemente para aldehídos aromáticos (pero no alifáticos) y se cree que el mecanismo involucra un dienolato quiral de metal.[40] [41]
Aldoles tiazoldinetiona criminas
Una versión más reciente de los auxiliares de Evans es la tiazoldinetiona crimina.[42] [43] Los rendimientos químicos, diastereselectividades y enantioselectividades de la reacción son generalmente altos, aunque no tan altos como en los casos Evans. Sin embargo, a diferencia de los auxiliares Evans, la tiazoldinetiona puede efectuar reacciones aldólicas acetato (ref: Crimmins, Org. Lett. 2007, 9(1), pp. 149–152.) y puede producir aductos "syn Evans" o "syn no-Evans" simplemente variando la cantidad de (-)-esparteina. Se cree que la reacción procede vía un estado de transición de seis miembros, con unión al titanio, análogo a los estados de transición propuestos para los auxiliares de Evans.
Reacciones aldólicas organocatalíticas
Otro desarrollo es el uso de catalizadores aminas secundarias quirales. Estas aminas secundarias forman enaminas transitorias al ser expuestas a cetonas, que pueden reaccionar enantioselectivamente con aldehídos electrófilos apropiados. Esto es conocido como catálisis enamina, un tipo de organocatálisis, dado que el catalizador está basado en una pequeña molécula orgánica. En un ejemplo seminal, la prolina catalizó eficientemente la ciclización de una tricetona:
Esta reacción es conocida como la reacción de Hajos-Parrish[44] [45] (también conocida como reacción de Hajos-Parrish-Eder-Sauer-Wiechert, refiriéndose a un informe contemporáneo de Schering de la reacción en condiciones más fuertes).[46] Bajo las condiciones de Hajos-Parrish, sólo se necesita una cantidad catalítica pequeña de prolina (3 mol%). No hay peligro de un fondo aquiral de la reacción porque los intermediarios transitorios de amina son más nucleofílicos que los enoles de las cetonas originarias. Esta estrategia es particularmente potente porque ofrece una forma simple de generar enantioselectividad en reacciones sin usar metales de transición, que tienen la posible desventaja de ser tóxicos o caros.
Es interesante el hecho de que las reacciones aldólicas catalizadas por prolina no muestran efectos no lineales (la enantioselectividad de los productos es directamente proporcional a la enantiopureza del catalizador). Combinado con evidencia de marcado isotópico y estudios computacionales, el mecanismo de reacción de las reacciones aldólicas catalizadas por prolina es como sigue:[47]
Esta estrategia permite la reacción de adición aldólica cruzada entre dos aldehídos, que de otro modo sería muy desafiante. En general, las reacciones aldólicas cruzadas entre aldehídos son desafiantes porque pueden polimerizarse fácilmente o reaccionar no selectivamente para dar mezclas estadísticas de los productos. El primer ejemplo se muestra a continuación:[48]
En contraste a la preferencia por aductos syn típicamente observada en las adiciones aldólicas basadas en enolato, estas adiciones aldólicas organocatalizadas son anti-selectivas. En muchos casos, las condiciones organocatalíticas son lo suficientemente suaves para evitar la polimerización. Sin embargo, la selectividad requiere una adición lenta y controlada del electrofílico deseado usando una jeringa, porque generalmente ambos reactivos son enolizables. Si un aldehído no tiene hidrógenos α enolizables, ni ramificaciones α o β, se puede lograr un control adicional.
Una demostración elegante del poder de las reacciones aldólicas asimétricas organocatalíticas fue expuesta por MacMillan y sus colaboradores en 2004, con su síntesis de carbohidratos diferencialmente protegidos. Mientras los métodos tradicionales de síntesis consiguen la síntesis de hexosas usando variaciones de estrategias de protección-deprotección iteractivas, requiriendo de 8 a 14 pasos, la organocatálisis permite acceder a muchos de los mismos sustratos usando un eficiente protocolo de dos pasos, involucrando la dimerización catalizada por prolina de α-oxialdehídos, seguido por ciclización aldólica de Mukaiyama en tándem.
La dimerización aldólica de α-oxialdehídos requiere que el aducto aldólico, en sí mismo un aldehído, sea inerte a reacciones aldólicas posteriores.[49] Estudios previos revelaron que los aldehídos que llevan un sustituyente α-alquiloxi o α-sililoxi son adecuados para esta reacción, mientras que los aldehídos que tienen grupos que atraen electrones como el acetoxi no eran reactivos. El producto eritrosa protegida puede ser luego convertida a los cuatro azúcares posibles vía la adición aldólica de Mukaiyama, seguida de formación de lactol. Esto requiere un diastereocontrol apropiado en la adición aldólica de Mukaiyama y el producto, un ion sililoxicarbenio, que preferentemente se cicle en vez de hacer una reacción aldólica posterior. Al final, se sintetizó glucosa, manosa y alosa:
Adiciones aldólicas "directas"
En una adición aldólica usual, un compuesto carbonílico se deprotona para formar un enolato. El enolato se agrega a un aldehído o cetona, lo que forma un alcóxido, que es protonado en un paso adicional. Un método superior, en principio, debería evitar la secuencia de deprotonación-aldol-protonación en favor de una "adición aldólica directa". La característica principal en tal proceso es que la adición aldólica genera un alcóxido, que es mucho más básico que los materiales iniciales, ocasionando la pérdida del catalizador:
Una aproximación demostrada por Evans es sililar el aducto aldólico:[50] [51]
Este método es más efectivo en cuanto a su coste e industrialmente útil que los procedimientos típicos basados en enolato. Una aproximación biomimética posterior elaborada por Shair usa β-tiocetoácidos como el nucleófilo.[52] El intermediario cetoácido es decarboxilado in situ (el ligante quiral como una bisoxazolina). Los aldehídos ramificados o aromáticos son sustratos pobres.
Véase también
- Reacción aldólica de Tishchenko
- Reacción de Baylis-Hillman
- Reacción de Ivanov
- Reacción de Reformatsky
- Reacción de Cannizzaro
- Ácido aldónico
- Condensación de Knoevenagel
Referencias
- ↑ Wade, L. G. (6th ed. 2005). Organic Chemistry. Upper Saddle River, New Jersey: Prentice Hall. pp. 1056–1066. ISBN 0-13-236731-9.
- ↑ Smith, M. B.; March, J. (5th ed. 2001). Advanced Organic Chemistry. New York: Wiley Interscience. pp. 1218–1223. ISBN 0-471-58589-0.
- ↑ Mahrwald, R. (2004). Modern Aldol Reactions, Volumes 1 and 2. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA. pp. 1218–1223. ISBN 3-527-30714-1.
- ↑ Heathcock, C. H. (1991). Comp. Org. Syn.. Oxford: Pergamon. pp. 133–179. ISBN 0-08-040593-2.
- ↑ Mukaiyama T. (1982). «The Directed Aldol Reaction». Org. React. 28: pp. 203–331.
- ↑ Paterson, I. (1988). «New Asymmetric Aldol Methodology Using Boron Enolates». Chem. Ind. 12: pp. 390–394.
- ↑ Wurtz, C. A. (1872). «.». Bull. Soc. Chim. Fr. 17: pp. 436–442.
- ↑ Wurtz, C. A. (1872). «Ueber einen Aldehyd-Alkohol». J. Prakt. Chemie 5 (1): pp. 457-464. doi:.
- ↑ Wurtz, C. A. (1872). «Sur un aldéhyde-alcool». Comp. Rend. 74: pp. 1361. http://gallica.bnf.fr/ark:/12148/bpt6k3031q/f1361.table.
- ↑ OCEANO, OCEANO. [www.oceano.com Diccionario Enciclopédico Interactivo] (2003 edición). Barcelona: GRUPO OCEANO. pp. 55-63. ISBN 84-9719-023-8. www.oceano.com. «...en 1872 fue descubierta independientemente por Alexander Porfyrevich Borodin.»
- ↑ Mestres R. (2004). «A green look at the aldol reaction». Green Chemistry 12: pp. 583–603. doi:.
- ↑ M. Braun, R. Devant (1984). «(R) and (S)-2-acetoxy-1,1,2-triphenylethanol - effective synthetic equivalents of a chiral acetate enolate». Tetrahedron Letters 25: pp. 5031–4. doi:.
- ↑ Jie Jack Li et al. (2004). Contemporary Drug Synthesis. Wiley-Interscience. pp. 118-. ISBN 0-471-21480-9.
- ↑ Schetter, B., Mahrwald, R. (2006). «Modern Aldol Methods for the Total Synthesis of Polyketides». Angew. Chem. Int. Ed. 45: pp. 7506–7525. doi:.
- ↑ Zimmerman, H. E.; Traxler, M. D. (1957). «The Stereochemistry of the Ivanov and Reformatsky Reactions. I». J. Am. Chem. Soc. 79: pp. 1920–1923. doi:.
- ↑ Heathcock C. H., Buse, C. T., Kleschnick W. A., Pirrung M. C., Sohn J. E., Lampe, J. (1980). «Acyclic stereoselection. 7. Stereoselective synthesis of 2-alkyl-3-hydroxy carbonyl compounds by aldol condensation». J. Org. Chem. 45: pp. 1066–1081. doi:.
- ↑ Bal, B.; Buse, C. T.; Smith, K.; Heathcock, C. H. Org. Syn., Coll. Vol. 7, p.185 (1990); Vol. 63, p.89 (1985). (Article)
- ↑ a b Brown H. C., Dhar R. K., Bakshi R. K., Pandiarajan P. K., Singaram B. (1989). «Major effect of the leaving group in dialkylboron chlorides and triflates in controlling the stereospecific conversion of ketones into either E- or Z-enol borinates». J. Am. Chem. Soc. 111: pp. 3441–3442. doi:.
- ↑ Ireland, R. E.; Willard, A. K. (1975). «The stereoselective generation of ester enolates». Tetrahedron Lett. 16 (46): pp. 3975–3978. doi:.
- ↑ Narula, A. S. (1981). «An analysis of the diastereomeric transition state interactions for the kinetic deprotonation of acyclic carbonyl derivatives with lithium diisopropylamide». Tetrahedron Lett. 22 (41): pp. 4119–4122. doi:.
- ↑ Ireland, R. E.; Wipf, P.; Armstrong, J. D. (1991). «Stereochemical control in the ester enolate Claisen rearrangement. 1. Stereoselectivity in silyl ketene acetal formation». J. Org. Chem. 56: pp. 650–657. doi:.
- ↑ Xie L., Isenberger K. M., Held G., Dahl, L. M. (1997). «Highly Stereoselective Kinetic Enolate Formation: Steric vs Electronic Effects». J. Org. Chem. 62: pp. 7516–7519. doi:.
- ↑ Evans D. A., Nelson J. V., Vogel E., Taber T. R. (1981). «Stereoselective aldol condensations via boron enolates». J. Am. Chem. Soc. 103: pp. 3099–3111. doi:.
- ↑ Evans D. A., Rieger D. L., Bilodeau M. T., Urpi F. (1991). «Stereoselective aldol reactions of chlorotitanium enolates. An efficient method for the assemblage of polypropionate-related synthons». J. Am. Chem. Soc. 113: pp. 1047–1049. doi:.
- ↑ Evans, D. A. et al. Top. Stereochem. 1982, 13, 1–115. (Review)
- ↑ Roush W. R. (1991). «Concerning the diastereofacial selectivity of the aldol reactions of .alpha.-methyl chiral aldehydes and lithium and boron propionate enolates». J. Org. Chem. 56: pp. 4151–4157. doi:.
- ↑ Masamune S., Ellingboe J. W., Choy W. (1982). «Aldol strategy: coordination of the lithium cation with an alkoxy substituent». J. Am. Chem. Soc. 104: pp. 1047–1049. doi:.
- ↑ a b Evans D. A., Dart M. J., Duffy J. L., Rieger D. L. (1995). «Double Stereodifferentiating Aldol Reactions. The Documentation of "Partially Matched" Aldol Bond Constructions in the Assemblage of Polypropionate Systems». J. Am. Chem. Soc. 117: pp. 9073–9074. doi:.
- ↑ Masamune S., Choy W., Petersen J. S., Sita L. R. (1985). «Double Asymmetric Synthesis and a New Strategy for Stereochemical Control in Organic Synthesis». Angew. Chem. Int. Ed. Engl. 24: pp. 1–30. doi:.
- ↑ Evans, D. A. Aldrichimica Acta 1982, 15, 23. (Review)
- ↑ Gage, J. R.; Evans, D. A. Organic Syntheses, Coll. Vol. 8, p.339 (1993); Vol. 68, p.83 (1990). (Article)
- ↑ Evans D. A., Bartroli J., Shih T. L. (1981). «Enantioselective aldol condensations. 2. Erythro-selective chiral aldol condensations via boron enolates». J. Am. Chem. Soc. 103: pp. 2127–2129. doi:.
- ↑ a b Evans D. A., Bender S. L., Morris J. (1988). «The total synthesis of the polyether antibiotic X-206». J. Am. Chem. Soc. 110: pp. 2506–2526. doi:.
- ↑ Evans D.A., Clark J.S., Metternich R., Sheppard G.S. (1990). «Diastereoselective aldol reactions using .beta.-keto imide derived enolates. A versatile approach to the assemblage of polypropionate systems». J. Am. Chem. Soc. 112: pp. 866–868. doi:.
- ↑ Evans D.A., Ng, H.P., Clark J.S., Rieger D.L. (1992). «Diastereoselective anti aldol reactions of chiral ethyl ketones. Enantioselective processes for the synthesis of polypropionate natural products». Tetrahedron 48: pp. 2127–2142. doi:.
- ↑ En esta reacción, el nucleófilo es un enolato de boro, derivado de la reacción con triflato de dibutilboro (nBu2BOTf), la base es N,N-Diisopropiletilamina. El tioéter se remueve en el paso 2 por reducción con hidrógeno / níquel Raney.
- ↑ Teruaki Mukaiyama, Kazuo Banno, and Koichi Narasaka (1974). «Reactions of silyl enol ethers with carbonyl compounds activated by titanium tetrachloride». J. Am. Chem. Soc. 96 (24): pp. 7503–7509. doi:.
- ↑ 3-Hydroxy-3-Methyl-1-Phenyl-1-Butanone by Crossed Aldol Reaction Teruaki Mukaiyama and Koichi Narasaka Organic Syntheses, Coll. Vol. 8, p.323 (1993); Vol. 65, p.6 (1987) Link
- ↑ Carreira E.M., Singer R.A., Lee W.S. (1994). «Catalytic, enantioselective aldol additions with methyl and ethyl acetate O-silyl enolates - a chira; tridentate chelate as a ligand for titanium(IV)». J. Am. Chem. Soc. 116: pp. 8837–8. doi:.
- ↑ Kruger J., Carreira E.M. (1998). «Apparent catalytic generation of chiral metal enolates: Enantioselective dienolate additions to aldehydes mediated by Tol-BINAP center Cu(II) fluoride complexes». J. Am. Chem. Soc. 120: pp. 837–8. doi:.
- ↑ Pagenkopf B.L., Kruger J., Stojanovic A., Carreira E.M. (1998). «Mechanistic insights into Cu-catalyzed asymmetric aldol reactions: Chemical and spectroscopic evidence for a metalloenolate intermediate». Angew. Chem. Intl. Ed. 37: pp. 3124–6. doi:.
- ↑ Crimmins M. T., King B. W., Tabet A. E. (1997). «Asymmetric Aldol Additions with Titanium Enolates of Acyloxazolidinethiones: Dependence of Selectivity on Amine Base and Lewis Acid Stoichiometry». Journal of the American Chemical Society 119 (33): pp. 7883–7884. doi:.
- ↑ Crimmins M. T., Chaudhary K. (2000). «Titanium enolates of thiazolidinethione chiral auxiliaries: Versatile tools for asymmetric aldol additions». Organic Letters 2 (6): pp. 775–777. doi:.
- ↑ Z. G. Hajos, D. R. Parrish, German Patent DE 2102623 1971
- ↑ Asymmetric synthesis of bicyclic intermediates of natural product chemistry Zoltan G. Hajos, David R. Parrish J. Org. Chem.; 1974; 39(12); 1615-1621. doi 10.1021/jo00925a003
- ↑ New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures Angewandte Chemie International Edition in English Volume 10, Issue 7, Date: July 1971, Pages: 496-497 Ulrich Eder, Gerhard Sauer, Rudolf Wiechert doi 10.1002/anie.197104961
- ↑ The ying and yang of asymmetric aminocatalysis Benjamin List Chem. Commun., 2006, 819–824, doi 10.1039/b514296m
- ↑ The First Direct and Enantioselective Cross-Aldol Reaction of Aldehydes Alan B. Northrup and David W. C. MacMillan J. Am. Chem. Soc.; 2002; 124(24) pp 6798–6799; (Communication) doi 10.1021/ja0262378
- ↑ Northrup A. B., Mangion I. K., Hettche F., MacMillan D. W. C. (2004). «Enantioselective Organocatalytic Direct Aldol Reactions of -Oxyaldehydes: Step One in a Two-Step Synthesis of Carbohydrates». Angewandte Chemie International Edition in English 43 (16): pp. 2152–2154. doi:.
- ↑ Diastereoselective Magnesium Halide-Catalyzed anti-Aldol Reactions of Chiral N-Acyloxazolidinones Evans, D. A.; Tedrow, J. S.; Shaw, J. T.; Downey, C. W. J. Am. Chem. Soc.; (Communication); 2002; 124(3); 392–393. doi 10.1021/ja0119548 10.1021/ja0119548; OL 2002, 4, 1127
- ↑ Evans, D. A.; Tedrow, J. S.; Shaw, J. T.; Downey, C. W. (2002). "Diastereoselective Magnesium Halide-Catalyzed anti-Aldol Reactions of Chiral N-Acyloxazolidinones". J. Am. Chem. Soc. 124 (3): 392–393. doi:10.1021/ja0119548
- ↑ Catalytic Enantioselective Thioester Aldol Reactions That Are Compatible with Protic Functional Groups Magdziak, D.; Lalic, G.; Lee, H. M.; Fortner, K. C.; Aloise, A. D.; Shair, M. D. J. Am. Chem. Soc.; (Communication); 2005; 127(20); 7284–7285. doi 10.1021/ja051759j
Categoría:- Reacciones químicas orgánicas
-













































Wikimedia foundation. 2010.