- Medicamento
-
 Medicamento en comprimidos.
Medicamento en comprimidos.
Un medicamento es uno o más fármacos, integrados en una forma farmacéutica, presentado para expendio y uso industrial o clínico, y destinado para su utilización en las personas o en los animales, dotado de propiedades que permitan el mejor efecto farmacológico de sus componentes con el fin de prevenir, aliviar o mejorar enfermedades, o para modificar estados fisiológicos.
Historia
Desde las más antiguas civilizaciones el hombre ha utilizado como forma de alcanzar mejoría en distintas enfermedades productos de origen vegetal, mineral, animal o en los últimos tiempos sintéticos.[1] El cuidado de la salud estaba en manos de personas que ejercen la doble función de médicos y farmacéuticos. Son en realidad médicos que preparan sus propios remedios curativos, llegando alguno de ellos a alcanzar un gran renombre en su época, como es el caso del griego Galeno (130-200). De él proviene el nombre de la Galénica, como la forma adecuada de preparar, dosificar y administrar los fármacos. En la cultura romana existían numerosas formas de administrar las sustancias utilizadas para curar enfermedades. Así, se utilizaban los electuarios como una mezcla de varios polvos de hierbas y raíces medicinales a los que se les añadía una porción de miel fresca. La miel además de ser la sustancia que sirve como vehículo de los principios activos, daba mejor sabor al preparado. En ocasiones se usaba azúcar. También se utilizaba un jarabe, el cual ya contenía azúcar disuelta, en vez de agua y el conjunto se preparaba formando una masa pastosa. Precisamente Galeno hizo famosa la gran triaca a la que dedicó una obra completa, y que consistía en un electuario que llegaba a contener más de 60 principios activos diferentes. Por la importancia de Galeno en la Edad Media, se hizo muy popular durante esta época dejando de estar autorizada para su uso en España en pleno siglo XX.[2]
Es precisamente en la Edad Media donde comienza su actividad el farmacéutico separado del médico. En su botica realiza sus preparaciones magistrales, entendidas como la preparación individualizada para cada paciente de los remedios prescritos, y se agrupan en gremios junto a los médicos. En el renacimiento se va produciendo una separación más clara de la actividad farmacéutica frente a médicos , cirujanos y especieros, mientras que se va produciendo una revolución en el conocimiento farmacéutico que se consolida como ciencia en la edad moderna. La formulación magistral es la base de la actividad farmacéutica conjuntamente con la formulación oficinal, debido al nacimiento y proliferación de farmacopeas y formularios, y esta situación continua hasta la segunda mitad del siglo XIX.[3]
A partir de este momento empiezan a aparecer los específicos, que consistían en medicamentos preparados industrialmente por laboratorios farmacéuticos. Es así, que las formas galénicas no adquirirán verdadero protagonismo hasta alrededor de 1940, cuando la industria farmacéutica se desarrolla y éstas comienzan a fabricarse en grandes cantidades. Desde entonces hasta hoy en día las maneras en que se presentan los medicamentos han evolucionado y la diversidad que encontramos en el mercado es muy amplia.[4]
Forma farmacéutica
Forma galénica o forma farmacéutica es la disposición individualizada a que se adaptan los fármacos (principios activos) y excipientes (materia farmacológicamente inactiva) para constituir un medicamento.[5] O dicho de otra forma, la disposición externa que se da a las sustancias medicamentosas para facilitar su administración.
El primer objetivo de las formas galénicas es normalizar la dosis de un medicamento, por ello también se las conoce como unidades posológicas. Al principio se elaboraron para poder establecer unidades que tuvieran una dosis fija de un fármaco con el que se pudiera tratar una determinada patología".[4]
La importancia de la forma farmacéutica reside en que determina la eficacia del medicamento, ya sea liberando el principio activo de manera lenta, o en su lugar de mayor eficiencia en el tejido blanco, evitar daños al paciente por interacción química, solubilizar sustancias insolubles, mejorar sabores, mejorar aspecto, etc.
Clasificación
Los medicamentos se dividen en cinco grupos:
- Especialidad farmacéutica: Es el medicamento de composición e información definidas, de forma farmacéutica y dosificación determinadas, preparado para su uso medicinal inmediato, dispuesto y acondicionado para su dispensación al público, con denominación, embalaje, envase y etiquetado uniformes según lo dispongan las autoridades sanitarias.
- Fórmula magistral: Es el medicamento destinado a un paciente individualizado, preparado por el farmacéutico, o bajo su dirección, para cumplimentar expresamente una prescripción facultativa detallada de las sustancias medicinales que incluye, según las normas técnicas y científicas del arte farmacéutico, dispensado en su farmacia o servicio farmacéutico y con la debida información al usuario.
- Preparado o fórmula oficinal: Es aquel medicamento elaborado y garantizado por un farmacéutico o bajo su dirección, dispensado en su oficina de farmacia o servicio farmacéutico, enumerado y descrito por el Formulario, destinado a la entrega directa a los enfermos a los que abastece dicha farmacia o servicio farmacéutico.
- Medicamento prefabricado: Es el medicamento que no se ajusta a la definición de especialidad farmacéutica y que se comercializa en una forma farmacéutica que puede utilizarse sin necesidad de tratamiento industrial y al que la autoridad farmacéutica otorgue autorización e inscriba en el registro correspondiente.
- Medicamento en investigación: Forma farmacéutica de una sustancia activa o placebo, que se investiga o se utiliza como referencia en un ensayo clínico, incluidos los productos con autorización de comercialización cuando se utilicen o combinen, en la formulación o en el envase, de forma diferente a la autorizada, o cuando se utilicen para tratar una indicación no autorizada, o para obtener más información sobre un uso autorizado.
Además, pueden recibir algunos calificativos específicos como:
- Medicamento citostático
- Medicamento compasivo
- Medicamento esencial
- Medicamento heroico: de acción muy enérgica que solo se aplica en casos extremos.[6]
- Medicamento huérfano
- Medicamento milagroso
- Medicamentos similares
Según la prescripción médica
En España y algunos países latinoamericanos, los medicamentos se dispensan, distribuyen o venden exclusivamente en las farmacias. Existen dos tipos de medicamentos según la prescripción médica:
- Medicamento de venta libre: Son aquellos medicamentos que se distribuyen libremente en las farmacias, sin necesidad de receta o prescripción médica. Se dividen en dos categorías:
- Las Especialidades farmacológicas publicitarias (EFP) se corresponden con medicamentos publicitados en los medios de comunicación de masas como, por ejemplo, la televisión.[cita requerida]
- Los productos OTC ("Over the Counter") son fármacos destinados al alivio, tratamiento o prevención de afecciones menores, con los que se posee una amplia experiencia de uso y han sido expresamente autorizados como tales.
- Medicamento con receta médica: Son aquellos medicamentos recetados por un médico para el tratamiento de una enfermedad o síntoma en concreto.
Según derecho de explotación
- Medicamento con patente: aquellos medicamentos de investigación propia del laboratorio que los comercializa, sujetos a la protección comercial que brindan las agencias internacionales de patentes.
La patente no se limita a la molécula, sino también a la formulación, mecanismo de producción, o asociación con otras moléculas. Mediante sucesión de patentes las casas farmacéuticas consiguen prolongar el periodo de exclusividad de sus presentaciones comerciales, aun cuando presentaciones anteriores de la misma molécula hayan quedado libres.
- Medicamento genérico: aquellas presentaciones de moléculas que ya no están protegidas por la patente de su investigador.
Pueden ser libremente producidas por otros laboratorios y suelen conllevar un menor precio. Las distintas Agencias del medicamento y organizaciones reguladores nacionales aseguran las similares bioequivalencia y biodisponibilidad de los medicamentos genéricos frente a aquellos que les son referencia.
Según la vía de administración
Existen numerosas formas de clasificar las formas galénicas, según el factor que tengamos en cuenta: su estado físico, la vía de administración, el origen de sus componentes, etcétera. No obstante la más utilizada y la más útil desde el punto de vista de la medicina es la clasificación según la vía de administración que usen.
Oral
La mayor parte de los fármacos administrados vía oral buscan una acción sistémica, tras un proceso previo de absorción entérica. En la absorción oral intervienen factores dependientes del individuo y otros dependientes de los fármacos que van a influir en la mayor o menor eficacia del fármaco administrado. Así mismo, la vía oral es motivo frecuente de interacciones farmacológicas, artículo éste que aconsejamos consultar para conocer la importancia de factores como el pH, toma o no de alimentos, tipo de éstos, velocidad del tránsito intestinal, u otros muchos que pueden influir en la absorción de un fármaco.
La vía oral constituye la vía más utilizada de administración de fármacos, subdividiéndose a su vez, en formas líquidas y formas sólidas.
A. Formas orales líquidas. No plantean problemas de disgregación o de disolución en el tubo digestivo, lo que condiciona una acción terapéutica más rápida. Por el contrario no están protegidas, en caso de reactividad, frente a los jugos digestivos. Resultan de elección particularmente en niños. Los líquidos para administración oral son habitualmente soluciones, emulsiones o suspensiones que contienen uno o más principios activos disueltos en un vehículo apropiado. Los vehículos pueden ser:
- Acuosos: sirven para disolver principios activos hidrosolubles. Los más comunes son los jarabes (que contienen una alta concentración de azúcar, hasta un 64% en peso).
- Mucílagos: líquidos viscosos resultantes de la dispersión de sustancias gomosas (goma arábiga, tragacanto, agar, metilcelulosa) en agua. Se usan, sobre todo, para preparar suspensiones y emulsiones.
- Hidroalcohólicos: los elixires son soluciones hidroalcohólicas (25% alcohol) edulcoradas utilizadas para disolver sustancias solubles en agua y alcohol.
Estas formas líquidas pueden contener también sustancias auxiliares para la conservación, estabilidad o el enmascaramiento del sabor del preparado farmacéutico (conservantes, antimicrobianos, antioxidantes, tampones, solubilizantes, estabilizantes, aromatizantes, edulcorantes y colorantes autorizados).
Las formas farmacéuticas líquidas para administración oral más usuales son:
Gotas
Son soluciones en las que el principio activo está concentrado.
Jarabes
Forma farmacéutica que consiste en una solución acuosa con alta concentración de carbohidratos tales como sacarosa, sorbitol, dextrosa, etc.; de consistencia viscosa, en la que se encuentra disuelto el o los principios activos y aditivos.[7]
Tisanas
Las tisanas son infusiones con baja concentración de principios activos.elaborada mediante la coccion o la infucion de una o varias hierbas medicinales.
Elixires
 Elixires de principios de siglo.
Elixires de principios de siglo.Forma farmacéutica que consiste en una solución hidroalcohólica, que contiene el o los principios activos y aditivos; contiene generalmente sustancias saborizantes, así como aromatizantes. El contenido de alcohol puede ser del 5 al 18 por ciento.[7]
Suspensiones
Son mezclas heterogéneas formadas por un sólido en polvo (soluto) o pequeñas partículas no solubles (fase dispersa) que se dispersan en un medio líquido (dispersante o dispersora). Sólido en líquido que no es soluble en este.
Suspensión extemporánea
Aquella que, por su poca estabilidad, se prepara en el momento de ser administrada.
Viales bebibles
 Comprimidos.
Comprimidos.B.Formas orales sólidas. Las formas sólidas, presentan una mayor estabilidad química debido a la ausencia de agua, lo que les confiere tiempos de reposición más largos. Además, estas formas galénicas permiten resolver posibles problemas de incompatibilidades, enmascarar sabores desagradables e incluso regular la liberación de los principios activos. Las formas farmacéuticas sólidas más frecuentes para administración oral son:
Comprimidos
Formas farmacéuticas sólidas que contienen, en cada unidad, uno o varios principios activos. Se obtienen aglomerando, por compresión, un volumen constante de partículas. Se administran generalmente por deglución, aunque se pueden dar otras posibilidades.
 Forma farmacéutica Cápsulas.
Forma farmacéutica Cápsulas.Cápsulas
Las cápsulas son preparaciones de consistencia sólida formadas por un receptáculo duro o blando, de forma y capacidad variable, que contienen una unidad posológica de medicamento. Este contenido puede ser de consistencia sólida, líquida o pastosa y estár constituido por uno o más principios activos, acompañados o nó de excipientes. El receptáculo se deshará por la acción de los jugos gástricos o entéricos, según la formulación, liberando entonces el principio activo.
Granulados
Agregados de partículas de polvos que incluyen principios activos, azucares y coadyuvantes diversos. Se presentan en forma de pequeños granos de grosor uniforme, forma irregular y más o menos porosidad. Existen granulados de distintos tipos: efervescentes, recubiertos, gastrorresistentes y de liberación modificada.
Sellos
Son cápsulas con un receptáculo de almidón. Prácticamente, han sido desplazados por las cápsulas duras.
Píldoras
Preparaciones sólidas y esféricas, destinadas a ser deglutidas íntegramente. Cada unidad contiene uno o más principios activos interpuestos en una masa plástica. Se encuentran en franco desuso habiendo sido desplazadas por los comprimidos y cápsulas.
Tabletas
Son pastillas para desleir en la cavidad bucal. Se diferencian de las píldoras por el tamaño y de los comprimidos por la técnica de elaboración. Sus constituyentes principales son la sacarosa, un aglutinante y uno o más principios activos.
Pastillas oficinales o trociscos
Presentan una consistencia semisólida y están constituidas primordialmente por los principios activos y goma arábiga como aglutinante. Suelen recubrirse, para su mejor conservación, con parafina o azúcar en polvo (escarchado). Se emplean para la vehiculización de antitusígenos y antisépticos pulmonares.
Liofilizados
Son preparaciones farmacéuticas que se acondicionan en forma de dosis unitarias y se liofilizan a continuación. Son formas muy porosas e hidrófilas y fácilmente dispersables en agua.
Colutorios
Son soluciones acuosas viscosas para el tratamiento tópico de afecciones bucales. se aplican con un pincel o una espátula. Estrictamente un colutorio se diferencia de un enjuague bucal al no llevar viscosizantes y puede ser sólido (polvos o comprimidos) para diluir antes de usar.[8]
Sublingual
Una forma especial de administración oral es la vía sublingual. En esta vía normalmente, se utilizan comprimidos que se disuelven debajo de la lengua absorbiéndose directamente. Tiene el inconveniente de ser exclusivamente permeable al paso de sustancias no iónicas, muy liposolubles. Esto hace que sólo puedan administrarse por esta vía fármacos de gran potencia terapéutica como la nitroglicerina o el isosorbide. Se utiliza para conseguir una acción terapéutica rápida o para fármacos que posean un alto grado de metabolización hepática, se degraden por el jugo gástrico o no sean absorbidos por vía oral. No obstante también se encuentran en el mercado presentaciones por comodidad del usuario. (Véase la formulación Flas en el epígrafe de innovaciones galénicas).
Parenteral
La biodisponibilidad de un fármaco administrado vía parenteral depende de sus características fisicoquímicas, de la forma farmacéutica y de las características anatomofisiológicas de la zona de inyección:
Vía intravenosa
Proporciona un efecto rápido del fármaco y una dosificación precisa, sin problemas de biodisponibilidad. Puede presentar, no obstante, graves inconvenientes, como la aparición de tromboflebitis, así como problemas de incompatibilidades entre dos principios activos administrados conjuntamente en la misma vía. Tiene el inconveniente de que no permite la administración de preparados oleosos debido a la posibilidad de originar una embolia grasa. Tampoco podrán usarse productos que contengan componentes capaces de precipitar algún componente sanguíneo o hemolizar los hematíes.
Vía intraarterial
Utilizada en el tratamiento quimioterápico de determinados cánceres; permite obtener una máxima concentración del fármaco en la zona tumoral, con unos mínimos efectos sistémicos.
Vía intramuscular
Se utiliza para fármacos no absorbibles por vía oral o ante la imposibilidad de administración del fármaco al paciente por otra vía ya que admite el ser utilizada para sustancias irritantes. Numerosos factores van a influir en la biodisponibilidad del fármaco por vía IM (vascularización de la zona de inyección, grado de ionización y liposolubilidad del fármaco, volumen de inyección, etc.). Esta vía es muy utilizada para la administración de preparados de absorción lenta y prolongada (preparados “depot”).
Vía subcutánea
De características similares a la anterior pero al ser la piel una zona menos vascularizada, la velocidad de absorción es mucho menor. Sin embargo, dicha velocidad puede ser incrementada o disminuida por distintos medios. No puede utilizarse para sustancias irritantes ya que podría producir necrosis del tejido.
Otras vías parenterales
De uso menos frecuente son la intradérmica, la intraaracnoidea o intratecal, epidural, intradural, intraósea, intraarticular, peritoneal o la intracardiaca.
Los preparados para administración parenteral son formulaciones estériles destinadas a ser inyectadas o implantadas en el cuerpo humano. A continuación se enumeran cinco de las más representativas:
- Preparaciones inyectables. Son preparaciones del principio activo disuelto (solución), emulsionado (emulsión) o disperso (dispersión) en agua o en un líquido no acuoso apropiado.
- Preparaciones para diluir previamente a la administración parenteral. Soluciones concentradas y estériles destinadas a ser inyectadas o administradas por perfusión tras ser diluidas en un líquido apropiado antes de su administración.
- Preparaciones inyectables para perfusión. Son soluciones acuosas o emulsiones de fase externa acuosa, exentas de pirógenos, estériles y, en la medida de lo posible, isotónicas con respecto a la sangre.
- Polvo para preparaciones inyectables extemporáneas. Sustancias sólidas estériles, dosificadas y acondicionadas en recipientes definidos que, rápidamente tras agitación, en presencia de un volumen prescrito de líquido estéril apropiado, dan lugar a soluciones prácticamente límpidas, exentas de partículas, o bien a suspensiones uniformes.
- Implantes o pellet. Pequeños comprimidos estériles de forma y tamaño adecuados que garantizan la liberación del principio activo a lo largo de un tiempo prolongado.
Por vía parenteral es posible la liberación retardada o prolongada de los principios activos a partir del punto de inyección. Esto se puede conseguir realizando diversas manipulaciones galénicas. Una de ellas consiste en sustituir una solución acuosa por una oleosa, en el caso de que el principio activo sea liposoluble. El método más clásico consiste en inyectar derivados poco hidrosolubles del principio activo, en forma de suspensiones amorfas o cristalinas (p. e., preparaciones retard de insulina). A veces, el principio activo puede también adsorberse sobre un soporte inerte desde el que será liberado, o bien fijarse en forma de microcápsulas, o incorporarse en liposomas para vectorizar algunos fármacos e, incluso ser tratado químicamente (profármaco) a fin de modificar sus propiedades fisicoquímicas.
Rectal
Supositorios
 Supositorios y antíguos moldes de fabricación.
Supositorios y antíguos moldes de fabricación.Son preparados de consistencia sólida y forma cónica y redondeada en un extremo. Tienen una longitud de 3-4 cm y un peso de entre 1-3 g. Cada unidad incluye uno o varios principios activos, incorporados en un excipiente que no debe ser irritante, el cual debe tener un punto de fusión inferior a 37 °C. Los excipientes de los supositorios pueden ser:
- Liposolubles: Son los más utilizados; entre ellos se encuentran la manteca de cacao, los glicéridos semisintéticos y los aceites polioxietilenados saturados.
- Hidrosolubles: polietilenglicoles (PEG).Hay muchas otras sustancias que se incluyen en ellos.[9] Hay dos tipos de supositorios, el primero de ellos es el supositorio de glicerogelatina y el segundo de ellos es el Polietilenglicol y los supositorios de PEG. Los supositorios PEG son buenos para los efectos a largo plazo del sistema, pero los supositorios de glicerogelatina son para los efectos locales y rápidos, ya que se disuelven rápidamente.
Cápsulas rectales
 Bolsa para aplicación de enema.
Bolsa para aplicación de enema.Soluciones y dispersiones rectales
- Enemas: Son formas galénicas líquidas, de composición variable, destinadas a ser administradas por vía rectal, empleando para ello dispositivos especiales. Pueden tener como objetivo la vehiculización de un principio activo (enemas medicamentosos), el vaciado de la ampolla rectal (enemas evacuantes) o el administrar una sustancia radio-opaca para la realización de estudios radiológicos (enemas opacos).
Pomadas rectales
La vía rectal puede utilizarse para conseguir efectos locales o sistémicos. En este último caso, sólo se debe considerar como una alternativa a la vía oral cuando ésta no pueda utilizarse, ya que la absorción por el recto es irregular, incompleta y además muchos fármacos producen irritación de la mucosa rectal. Uno de los pocos ejemplos en los que esta forma farmacéutica tiene una indicación preferente es el tratamiento de las crisis convulsivas en niños pequeños (diacepam).
También existen pomadas rectales para el tratamiento de las hemorroides externas.Tópica
La vía tópica utiliza la piel y las mucosas para la administración de fármaco. Así pues, esto incluye mucosa ocular y genital. La mucosa oral ya ha sido vista dentro del epígrafe de la vía oral. La característica de esta vía es que se busca fundamentalmente el efecto a nivel local, no interesando la absorción de los principios activos. Los excipientes fundamentales para las formulaciones galénicas de uso tópico son tres: líquidos, polvos y grasas. Estos pueden combinarse entre sí de numerosas formas para adaptarse a las características del sitio en donde se van a aplicar, de ahí la variedad de formas galénicas para uso tópico.[10]
POLVOS Loción agitable (Líquido + Polvo) LÍQUIDO Pasta al agua (Líquido+Polvo+Grasa) Pasta (Grasa+Polvo)
GRASA Crema (Grasa+Líquido)
Tabla de tres entradas, con las posibles combinaciones entre Grasas, líquidos y polvos.[10] Baños
Consisten en la inmersión de todo o parte del cuerpo en una solución acuosa a la que se añaden determinados productos. Los más utilizados son los baños coloidales, que tanto tibios como calientes actúan como sedantes y antipruriginosos. En su preparación se mezcla una taza de almidón en un litro de agua y posteriormente esta preparación se hecha al agua del baño. En los baños oleosos se sustituye el almidón por aceites fácilmente dispersables, produciendo una suspensión homogénea. Actualmente se utilizan sobre todo los baños de sales, potenciados por el mundo de la cosmética.
Lociones

También conocidas en dermatología como curas húmedas abiertas son muy útiles en inflamaciones superficiales como el eccema. Se colocan compresas empapadas en ciertos líquidos en las zonas secretantes y se cambian cada 5-10 minutos, o bien se añade de forma periódica líquido, de tal manera que se evite la desecación por efecto del calor de la piel. Esto se suele mantener durante 2 horas tres veces al día. El objetivo es el enfriamiento que consigue la evaporación del líquido, que actúa como antiinflamatorio. Además tienen propiedades antipruriginosas y vasoconstrictoras. Jamás irritan y no tienen riesgos en su uso. Los líquidos utilizados pueden ser el suero fisiológico, etanol al 10%, el permanganato potásico al 0.01% o la solución de Burow, compuesta por agua, subacetato de plomo y sulfato alumínico potásico. Una loción clásica de la farmacopea española sería el Agua de Dalibour, compuesta por una solución hidroalcohólica al 1% y sulfato de cobre, sulfato de cinc y alcanfor e indicada en el tratamiento del impétigo, dermatitis sobreinfectadas y dermatosis exudativas.[11]
Toques o pincelaciones
Se suelen utilizar para descostrar heridas o lesiones dérmicas. Se utiliza el etanol al 10% al que se le puede añadir glicerina al 5%. El etanol puede causar quemazón, por lo que se desaconseja en región genital y cara. Una forma especial serían las pincelaciones secantes de la farmacopea alemana, que son en realidad lociones agitables, constituidas a partes iguales en peso de polvos y liquido, aunque son algo más cosméticas con una proporción algo inferior de polvos. A estas lociones se les puede añadir principos activos en forma de líquido o de polvo. Las indicaciones son las mismas que las de los polvos (ver después), ya que en realidad lo que se busca es el depósito de éstos sobre la piel al evaporarse el líquido de la formulación.[10]
Pinturas
Son preparaciones líquidas, coloreadas (de donde su nombre), resultado de la mezcla de hidroalcoholes con drogas secas a temperatura ambiente. Sus propiedades dependen del fármaco añadido, aunque en lineas generales son antisépticas y antipruriginosas. Lo más habitual suele ser el uso de soluciones de etanol, éter o cloroformo con productos como el mentol, fenol, fucsina, eosina, violeta de genciana o breas. Si resecan demasiado se puede añadir aceite de ricino al 1%. Se suelen administrar una vez al día y se retiran los restos al día siguiente antes de la nueva aplicación.
Linimentos
Forma farmacéutica que consiste en una presentación líquida, solución o emulsión que contiene el o los principios activos y aditivos cuyo vehículo es acuoso, alcohólico u oleoso, y se aplica exteriormente en fricciones.
Polvos
Los polvos se obtienen por división sucesiva de productos sólidos y secos hasta partículas homogéneas de tamaño variable. Tras su fabricación son pasados por tamices que oscilan entre los 0.1 mm de apertura del enrejado (polvos muy finos) hasta 1 mm. (polvos gruesos). Aplicados sobre la piel forman una capa finísima de propiedades refrescantes, vasoconstrictoras, antiinflamatorias y antipruriginosas. También protegen del roce entre superficies (pliegues) y ejercen protección mecánica. Los más famosos son los polvos de talco, que en dermatología se pueden asociar a otras sustancias como el óxido de zinc. También se cuenta con polvos de origen vegetal (almidón, licopodio), y en la industria cosmética se han utilizado ampliamente añadiendo pigmentos inócuos, del tipo del cinabrio, eosina, bentonita, óxido de hierro o ictiol para pieles oscuras. Aunque se pueden fabricar polvos directamente con las propias sustancias activas, no es esto lo más recomendable, siendo mejor su incorporación a los polvos inertes citados anteriormente. Así, se pueden añadir sustancias del tipo de las sulfamidas, fungicidas (ciclopirox olamina, miconazol), anhidróticos (cloruro de aluminio), etc. Como inconveniente importante tiene el que no se deben de utilizar ante presencia de secreciones, pues la mezcla con el exudado o el pus forma una especie de cemento que favorece la infección, es irritante y dificulta la cicatrización.[12] Es especialmente desaconsejable la extendida utilización de polvos asociados a productos antipruriginosos en la varicela, donde pueden dejar cicatrices más intensas de lo habitual o la utilización de polvos de boro y ácido salicílico, muy extendidos por parte de la industria farmacéutica y de efectos no demostrados.[13]
Pastas
Están compuestas por polvos y grasas a partes iguales, aunque si son demasiado espesas se puede reducir la parte correspondiente a los polvos, aumentando la cantidad correspodiente de grasa. En líneas generales las pastas son secantes, pero mantienen la piel suave y plegable, protegiendo de traumas mecánicos y permitiendo la transpiración. Están indicadas en procesos secos (dermatitis crónicas) o muy poco secretantes y en la prevención de las úlceras de decúbito, por su efecto protector. Pueden incorporar principios activos tanto en forma líquida como sólida. Sus contraindicaciónes principales son las lesiones secretantes, infecciones y regiones pilosas. Se aplican extendiéndolas con una espátula de madera y se retiran con aceite de oliva o vaselina líquida. En lugar de grasas sólidas se pueden usar aceites, dando lugar a pastas oleosas. Si a una pasta se le añaden líquidos (agua), nos encontramos ante la pasta acuosa o pasta refrescante.
Pomadas o ungüentos
Son grasas o sustancias de parecidas características que presenten aspecto semisólido a 25 °C. Es esta propiedad física lo que realmente las define ya que la composición química es enormemente variada.[10] El término pomada es habitualmente usado en la literatura hispánica, mientras que el término ungüento es de uso preferente en el ámbito anglosajón, aunque por efecto de la preponderancia científica, éste se va extendiendo como forma principal de uso. Entre no entendidos se aplica el término pomada a cualquier presentación sólida de aplicación en la piel, sea crema, gel o pomada, de la misma forma que llaman pastilla a la mayoría de las formas galénicas de administración oral. Sus indicaciones son amplias: mejoran la piel seca y agrietada, son excelentes para retirar costras o escamas y son medianamente toleradas en zonas pilosas. Admiten multitud de principios activos bien en solución, los principios activos liposolubles, bien en forma de polvo o en dispersión coloidal para los insolubles en grasas. Su contraindicación más importante son los procesos inflamatorios agudos, tanto más contraindicada cuanto más húmedo sea el proceso.
Emulsiones
 Gel para el cabello.
Gel para el cabello.Una emulsión es una mezcla más o menos homogénea de dos liquidos inmiscibles. Un líquido (la fase dispersa) es dispersado en otro (la fase continua o fase dispersante). Muchas emulsiones son emulsiones de aceite y agua.
Geles
Respecto a los anteriores es de creación mucho posterior. Son emulsiones semisólidas de polímeros orgánicos (metilcelulosa, agar, gelatina, propilenglicol, galato de propilo, edetato disódico, carboxipolímero) en un líquido (agua).[14] Se suele añadir hidróxido sódico o ácido clorhídrico para ajustar el pH. Son, pues, bases incoloras, claras, no grasas, miscibles con agua. Se pueden considerar como un sistema coloidal donde la fase continua es sólida y la dispersa es líquida. En esta fase líquida puede incorporar principios activos en solución. A temperatura ambiente son sólidos o semisólidos, fluidificándose al ser calentadas. Por su aspecto cosmético se suelen utilizar en zonas pilosas o estrechas (conducto auditivo externo, fosas nasales). Tras su aplicación desaparecen rápida y completamente.
Champús
 Botellas de champú y lociones faciales de principios del siglo XX.
Botellas de champú y lociones faciales de principios del siglo XX.Forma galénica utilizada para el cuidado del cabello y del cuero cabelludo.
Colirios
Forma farmacéutica que consiste en una solución que contiene el o los principios activos y aditivos, aplicable únicamente a la conjuntiva ocular. Debe ser totalmente transparente, estéril, isotónica y con un pH neutro o cercano a la neutralidad.[15]
Gotas óticas y nasales
Las gotas óticas y nasales son una forma galénica para uso tópico consistente en un líquido acuoso u oleoso en el que van incorporados los principios activos y que se utilizan en el conducto auditivo externo o las fosas nasales respectivamente. Sus propiedades son similares a las de los colirios, salvo la exigencia de la esterilidad.[15] Acorde a la gran cantidad de principios activos que tolera esta forma galénica, las indicaciones también son amplias:
- Procesos infecciosos: Otitis externa, otitis de las piscinas, ectima nasal, etc.
- Procesos alérgicos, para el caso de las gotas nasales. La rinitis alérgica es una patología de alta prevalencia que en muchas ocasiones no es controlable con tratamiento sistémico.
- Procesos inflamatorios nasales (polipos nasales) u óticos.
- Patologías dermatológicas que afectan la piel del oido, como el psoriasis.
Respecto a las contraindicaciones habrá que tener en cuenta las siguientes consideraciones:
- Gotas nasales: Evitar sustancias que alteren la actividad de los cilios de las células de la mucosa nasal, como las sustancias boricadas.
- Gotas óticas: En lo posible evitar las soluciones oleosas y tener presente la posibilidad de que la membrana timpánica no esté indemne.
Apósitos
Cualquiera de los diferentes productos sanitarios empleados para cubrir y proteger una herida.[16] Su finalidad última es la de promover la cicatrización de la herida. En función a ello, las características que ha de reunir un buen apósito son:[17]
- Impermeabilidad a los gérmenes, partículas y agua.
- Capacidad de absorción.
- Favorecimiento del pH ácido.
- Esterilidad.
- Permeabilidad a los gases.
- Proporcionar aislamiento térmico.[18]
A estas propiedades habría que añadirles otras derivadas de la interacción con el organismo:[19]
- Elasticidad y flexibilidad.
- Baja adherencia a la herida.
- Alto grado de cohesión.
- No tóxico.
- No alergizante.
Percutánea o transdérmica
A pesar de lo comentado respecto a la dificultad para la absorción de los principios activos, siempre existe la posibilidad de que se absorba una parte de los mismos a través de la piel. Este fenómeno se da con más intensidad en las zonas donde la piel está menos queratinizada (cara), donde se favorece la humedad (pliegues corporales) o en pieles más sensibles como la de los niños. Por otra parte, en ocasiones, sí que nos interesa que el principio activo sea absorbido para hacer su efecto a nivel sistémico. Esta vía, se conoce como vía percutánea o vía transdérmica y presenta unas características especiales.
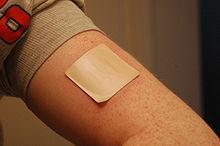 Parche transdérmico aplicado en el brazo.
Parche transdérmico aplicado en el brazo.Los sistemas terapéuticos transdérmicos (STT) son formas de dosificación ideados para conseguir el aporte percutáneo de principios activos a una velocidad programada, o durante un periodo de tiempo establecido. Existen varios tipos de sistemas transdérmicos, entre los que se encuentran:
Parches transdérmicos
Forma galénica consistente en un reservorio con principio activo que se libera lentamente al aplicarlo sobre la piel. Se persigue que el fármaco pase a la circulación sistémica a través de la piel y no la actividad del fármaco en la propia piel. Estos parches proporcionan niveles plasmáticos terapéuticos constantes del fármaco, siempre que la piel permanezca intacta.[20] Aunque relativamente recientes,[21] tienen numerosas indicaciones y son objeto de continua investigación.[22] ,[23] ,[24] ,[20]
La iontoforesis
Técnica utilizada en fisioterapia que consiste en la introducción de un fármaco o principio activo (mediante la utilización de corriente galvánica u otras formas de corrientes derivadas de esta) a través de la piel.
Consiste en la colocación sobre la piel de dos electrodos que, por su polaridad, hacen que un fármaco cargado iónicamente atraviese la piel. Se produce por la interacción de la polaridad del electrodo con la carga iónica de la sustancia elegida (mediante el rechazo de los iones en el polo del mismo signo).
Las vías de introducción a través de la piel son sobre todo: folículos pilosos, glándulas sudoríparas y sebáceas, por interponer menor resistencia para el paso de sustancias.
De esta manera son administrados por vía percutánea fármacos cargados eléctricamente, sin producir efectos generales en el organismo, ya que sus efectos son locales.
Inhalada
 Presentación clásica de un aerosol.
Presentación clásica de un aerosol.- Aerosoles: son dispositivos que contienen soluciones o suspensiones de un principio activo, envasadas en un sistema a presión de manera que, al accionar la válvula, se produce la liberación del principio activo en forma de microgotas impulsado gracias a un agente propelente. Los principios activos que pueden ser vehiculizados en esta forma galénica son numerosos:
-
- Agonistas β2 adrenérgicos:
- De Acción corta: salbutamol, levalbuterol, terbutalina y bitolterol.
- De acción prolongada: salmeterol, formoterol, bambuterol.
- Anticolinérgicos, como el bromuro de ipratropio.
- Glucocorticoides: beclometasona o fluticasona.
Su uso por via inhalada nos orienta hacia las patologías que se beneficiarían del tratamiento con los aerosoles:[25]
-
- Crisis asmática.
- Tratamiento de mantenimiento de la alergia respiratoria o asma extrínseca.
- En la enfermedad pulmonar obstructiva crónica (EPOC) tanto en el mantenimiento como en la profilaxis de las reagudizaciones.
 Sistema de nebulización por compresión.
Sistema de nebulización por compresión.
- Inhaladores de polvo seco: a partir del medicamento en estado sólido, se liberan partículas suficientemente pequeñas de forma sincrónica con la inspiración; la fuerza de la inhalación arrastra el producto.
- Nebulizadores: son dispositivos que hacen pasar una corriente de aire sobre un líquido que lleva el principio activo en disolución. Se generan partículas uniformes y muy finas del principio activo (líquido) que son inspiradas junto con el aire que inyecta el nebulizador. Este sistema permite que el fármaco penetre más profundamente en las vías aéreas.
- Inhalaciones.
- Insuflaciones.
Innovaciones galénicas
La industria farmacéutica está continuamente desarrollando nuevas formas de hacer llegar el fármaco a su destino de la forma más rápida y eficaz. Al igual que el descubrimiento de nuevos fármacos, las nuevas galénicas están sujetas a patente de propiedad, pasando al cabo de un tiempo a dominio público. Así, por ejemplo, Norman Leo Henderson y Louis Nasir Elowe inventaron en 1969 las cápsulas de liberación prolongada con la patente 3427378.[26] De esta manera, continuamente se están inventando sistemas, que basados en las fórmulas galénicas clásicas, aportan un plus de efectividad al producto comercial. Algunos ejemplos serían:
- Sistema OROS o de Microbomba osmótica. Es un sistemas que permite la liberación temporal controlada del fármaco. Está constituido por un reservorio que contiene el fármaco, formado por un núcleo sólido con capacidad osmótica. Rodeando el reservorio existe una membrana semipermeable que permite el paso del agua procedente del exterior del sistema. Cuando el comprimido entra en contacto con el jugo gastrointestinal, la penetración del agua produce la disolución del núcleo osmótico y la salida del medicamento por un orificio o zona de liberación. El tamaño del poro de la membrana semipermeable va a condicionar la mayor o menor entrada de agua y, por tanto, la velocidad de liberación del principio activo.[27]
- Sistema Filmtab.
- Sistema MUPS : En 1998 AstraZéneca desarrolló el sistema MUPS (Multiple Unit Pellet System) o Sistema Multigranular. La formulación de los comprimidos MUPS permite una liberación rápida de 1.000 a 2.000 unidades de principio activo con protección frente al ácido en el estómago. Son más pequeñas que las unidades contenidas en una cápsula tradicional, se dispersan con facilidad y se disuelven en el intestino delgadoofreciendo una eficacia mas predecible.[28]
- Liposomas: Los liposomas son vesículas extraordinariamente pequeñas compuestas principalmente por fosfolípidos organizados en bicapas. Estas vesículas contienen una fase acuosa interna y están suspendidas en una fase acuosa externa. Se utilizan básicamente para transportar los principios activos de una manera lo más selectiva posible. Dependiendo de su naturaleza, el fármaco se puede incorporar dentro del liposoma (si es hidrofílico) o en la bicapa liposomal (caso de los lipofílicos).[29] Las ventajas de esta forma galénica serían:
- Aumento de la eficacia y disminución de la toxicidad del principio activo encapsulado.
- Prolongación del efecto.
- Mejor absorción, penetración y difusión.
- Posibilidad de vías de administración alternativas.
- Estabilización del principio activo.
- Sistema Flas: "Es una forma sólida oral que tiene como propiedad que no hace falta deglutir el contenido, sino que se absorbe directamente en contacto con la cavidad bucal"[4]
- Sistema Chronosphère.
- Gel Termoreversible: forma farmacéutica que permite que la preparación se mantenga en forma de solución líquida a temperaturas inferiores a 25 °C; en cambio a temperaturas próximas a 35 °C (cuando entra en contacto con el cuerpo humano) aumenta su viscosidad y la solución líquida se transforma en gel. Es ideal para uso en mucosas, ya que la forma líquida favorece la penetración pero al convertirse en sólido se evita el goteo y la deglución. Los laboratorios españoles SALVAT utilizan este sistema para algunos de sus productos de uso por vía nasal. [11]
Producción
Los medicamentos son producidos generalmente por la industria farmacéutica. Los nuevos medicamentos pueden ser patentados, cuando la empresa farmacéutica ha sido la que ha investigado y lanzado al mercado el nuevo fármaco. Los derechos de producción o licencia de cada nuevo medicamento está limitado a un lapso que oscila entre 10 y 20 años. Los medicamentos que no están patentados se llaman medicamentos copia; en cambio, aquellos que no están patentados pero tienen un estudio de bioequivalencia, aprobado por las autoridades locales, se llaman medicamentos genéricos.
Estudios de estabilidad
El objetivo de estos estudios es proveer evidencia documentada de como las características físicas, químicas, microbiológicas y biológica del medicamento varían con el tiempo bajo la influencia de factores como temperatura, humedad, luz y establecer las condiciones de almacenamiento adecuadas y el periodo de caducidad.
La estabilidad de un fármaco se puede definie como la propiedad de éste envasasado en un determinado material para mantener durante el tiempo de almacenamiento y uso de las características físicas, químicas, fisicoquímicas, microbiológicas y biológicas entre los límites especificados.
Las formas de inestabilidad de un fármaco son:
- Química
Solvólosis, oxidación, deshidratación, racemización, incompatibilidades de grupo.
- Física
Polimorfismo, vaporización, adsorción.
- Biológica
Fermentación y generación de toxinas
Fechas de expiración
En el año 1979, en Estados Unidos se establecio una ley que pedia a las empresas productoras de medicamentos que pusieran la fecha hasta la cual se garantizaba la maxima potencia del medicamento, en otras palabras, no indica el momento en que yo es seguro tomarlas ni cuando se vuelven dañinas. La mayoria de medicinas son muy durables y se degradan lentamente, por lo que la mayoria, mientras haya sido apropiadamente fabricada, mantienen su potencia en altos rangos muy despues de su fecha de caducación. Un ejecutivo de Bayer, Chris Allen, dijo que las aspirinas que fabrican llevan una fecha de caducidad a los tres años luego de su fabricación, pero esta podria ser de incluso más y esta aun estaria en su maxima potencia, el motivo detras de esto yace en que constantemente estan mejorando la formula de las aspirinas, y hacer pruebas de caducidad de más de 4 años por cada actualización seria irrazonable. Sin embargo, se han registrado problemas con medicamentos que incluyen tetraciclina; también hay excepciones en la duración, como con nitroglicerina, insulina, y algunos antibioticos líquidos.[30] [31] Los fármacos se administran con el fin de conseguir un objetivo terapéutico
La concentración adecuada y la dosis requerida para alcanzar este objetivo dependen, entre otros factores, del estado clínico del paciente, la gravedad de la patología a tratar, la presencia de otros fármacos y de enfermedades intercurrentes.
Para ello se requiere no solamente lograr una respuesta farmacológica y poder mantenerla; por lo tanto, es necesario alcanzar la concentración apropiada del fármaco en el lugar de acción. Para ello es necesario conocer su farmacocinética.
Debido a las diferencias individuales, el tratamiento eficaz requiere planificar la administración según las necesidades del paciente.
Tradicionalmente, esto se efectuaba por medio del ajuste empírico de la dosis hasta conseguir el objetivo terapéutico.
Sin embargo, a menudo este método es poco adecuado debido a la demora en conseguir el efecto o a la aparición de toxicidad. Una aproximación alternativa consiste en iniciar la administración de acuerdo con la absorción, distribución y eliminación, esperadas en el paciente y, posteriormente, ajustar la dosis según la respuesta clínica y por medio del monitoreo de las concentraciones plasmáticas.
Este enfoque requiere conocer la farmacocinética del fármaco en función de la edad, el peso y las consecuencias cinéticas de las posibles enfermedades intercurrentes (renales, hepáticas, cardiovasculares o una combinación de ellas).
PARÁMETROS FARMACOCINÉTICOS BÁSICOS
El comportamiento farmacocinético de la mayoría de los fármacos puede resumirse por medio de algunos parámetros. Los parámetros son constantes, aunque sus valores pueden diferir de un paciente a otro y, en el mismo enfermo, en situaciones distintas.
La biodisponibilidad expresa el grado de absorción por la circulación sistémica.
La constante de absorción define la velocidad de absorción.
Las modificaciones de estos dos parámetros influyen sobre la concentración máxima o pico, el tiempo que tarda en alcanzarse la concentración máxima y el área bajo la curva (ABC) concentración-tiempo tras una dosis oral única.
En los tratamientos crónicos, el grado de absorción es la medida más importante por la que se relaciona con la concentración media, mientras que el grado de fluctuación depende de la constante de absorción.
El volumen aparente de distribución es el líquido teórico corporal en que tendría que haberse disuelto el fármaco para alcanzar la misma concentración que en el plasma. Se usa para saber la dosis requerida para alcanzar una concentración determinada en la sangre. La fracción libre es útil porque relaciona la concentración total con la libre, que es quién, presumiblemente, está más asociada con los efectos farmacológicos. Es un parámetro útil sobre todo si se altera la fijación a las proteínas plasmáticas, por ejemplo en caso de hipoalbuminemia, de enfermedad renal y/ó hepática y en interacciones por desplazamiento de la unión a dichas proteínas. El volumen aparente de distribución y la fracción libre son los parámetros más utilizados para el estudio de la distribución del fármaco.
La velocidad de eliminación de un fármaco del organismo es proporcional a su concentración plasmática. El parámetro que relaciona a ambas medidas es el aclaramiento total o clearance, que es la suma del aclaramiento renal más el aclaramiento extrarrenal o metabólico.
La fracción de fármaco eliminado sin cambios (en forma inalterada) es un parámetro útil para evaluar el efecto potencial de las enfermedades renales y hepáticas sobre la eliminación de los fármacos. Una fracción baja indica que el metabolismo hepático es el mecanismo de eliminación y que una enfermedad hepática podría afectar la eliminación del fármaco. Las patologías renales producen mayores efectos en la cinética de fármacos con elevada fracción de fármaco eliminado inalteradamente.
La velocidad con que se extrae un fármaco de la sangre por un órgano excretor como el hígado no puede exceder la velocidad a la que llega a dicho órgano. Es decir, el aclaramiento presenta un valor límite. Cuando la extracción es elevada, la eliminación está limitada por la llegada de fármaco al tejido y, por tanto, por la perfusión de éste. Cuando el órgano de eliminación es el hígado o la pared intestinal y el fármaco se administra vía oral, una porción de la dosis administrada puede ser metabolizada durante su paso obligado a través de los tejidos hasta la circulación sistémica; es lo que se denomina efecto o metabolismo de primer paso hepático.
Por tanto, siempre que una sustancia tenga una extracción (aclaramiento) elevada en hígado o pared intestinal, la biodisponibilidad por vía oral, será baja, hasta el punto que a veces puede desaconsejar la administración vía oral,. o requerir la administración de una dosis oral muy superior a la dosis parenteral equivalente.
Algunos ejemplos de fármacos presentan un efecto de primer paso importante;
ð alprenolol,ð hidralazina,
ð isoproterenol,
ð lidocaína,
ð meperidina,
ð morfina,
ð nifedipina,
ð nitroglicerina,
ð propranolol,
ð testosterona
ð verapamilo.
La constante de eliminación está en función de cómo se elimina el fármaco de la sangre por parte de los órganos excretores y cómo se distribuye por el organismo.
La vida media de eliminación beta, (o semivida) es el tiempo requerido para que la concentración plasmática -o la cantidad de fármaco del organismo- se reduzca en un 50%.
Para la mayoría de los fármacos, la vida media se mantiene constante a pesar de la cantidad de fármaco que esté presente en el organismo, aunque existen excepciones (difenilhidantoína, teofilina y heparina).
Tiempo medio de permanencia (TMP) es otra medida de la eliminación del fármaco. Se define como el tiempo medio que la molécula de un fármaco permanece en el organismo tras una administración intravenosa rápida. Al igual que el aclaramiento, su valor es independiente de la dosis administrada. Para fármacos que describen un modelo de distribución monocompartimental, el TMP se iguala al recíproco de la constante de eliminación.
VARIABILIDAD DE LOS PARÁMETROS FARMACOCINÉTICOS
Se han identificado muchas de las variables que afectan los parámetros farmacocinéticos, las cuales deben tenerse en cuenta para ajustar la dosis administrada a las necesidades de cada paciente. Dado que incluso después del ajuste de las dosis suele persistir una notable variabilidad, es necesaria una monitorización cuidadosa de la respuesta farmacológica y, en muchos casos, de la concentración plasmática.
Edad y peso.
Para algunos fármacos se han establecido con precisión los cambios farmacocinéticos relacionados con la edad y el peso. En los niños y jóvenes (de 6 meses a 20 años), la función renal se correlaciona bien con la superficie corporal. Así, en el caso de los fármacos que se eliminan principalmente en forma inalterada por la orina, el aclaramiento varía con la edad de acuerdo con el cambio de la superficie corporal. En las personas mayores de 20 años, la función renal disminuye aproximadamente un 1% al año. Teniendo en cuenta estos cambios, es posible ajustar la dosis de dichos fármacos en cada edad. También se ha demostrado que la superficie corporal se correlaciona con el aclaramiento metabólico en los niños, aunque hay muchas excepciones.
En los recién nacidos y los lactantes, tanto la función renal como la hepática no están completamente desarrolladas y no es posible hacer generalizaciones, excepto que existen cambios rápidos.
Alteración de la función renal
El aclaramiento renal de la mayoría de los fármacos varía proporcionalmente al aclaramiento de creatinina, cualquiera que sea la patología renal. El cambio en el aclaramiento total depende de la contribución de los riñones en la eliminación total de dicho fármaco. Por tanto, se espera que el aclaramiento total sea proporcional a la función renal (aclaramiento de creatinina) para fármacos que se excretan exclusivamente de manera inalterada y no resulte afectado en el caso de fármacos que se eliminan por metabolismo.
A veces, en la insuficiencia renal se modifica el volumen aparente de distribución. En el caso de la digoxina, la reducción del volumen de distribución depende de la menor fijación a los tejidos. En el caso de la difenilhidantoína, el ácido salicílico y muchos otros fármacos, el volumen de distribución aumenta debido a que se reduce la fijación a las proteínas plasmáticas.
En las situaciones de estrés fisiológico (p.ej.,infarto de miocardio, cirugía, colitis ulcerosa o enfermedad de Crohn) aumenta la concentración del reactante de fase aguda (1-glucoproteína ácida). En consecuencia, aumenta la fijación a las proteínas por parte de algunos fármacos de naturaleza básica como el propranolol, la quinidina y la disopiramida; por consiguiente, disminuye el volumen aparente de distribución de estos fármacos.
Las enfermedades hepáticas producen modificaciones en el aclaramiento metabólico, pero no se dispone de buenos predictores de estos cambios. Se ha descrito una reducción espectacular de la metabolización de fármacos en la cirrosis hepática. En esta enfermedad a menudo se observa una disminución de la fijación a las proteínas plasmáticas debido a la menor concentración de albúmina en sangre. Habitualmente, la hepatitis aguda cursa con elevación de las enzimas séricas, por eso no se asocia a una alteración del metabolismo.
Otras enfermedades como la insuficiencia cardíaca, la neumonía, el hipertiroidismo y muchas otras enfermedades también pueden alterar la farmacocinética.
Interacciones farmacológicas.
Las interacciones farmacológicas provocan cambios en los valores de los parámetros farmacocinéticos y, por tanto, en la respuesta terapéutica. Se sabe que las interacciones afectan todos los parámetros. La mayoría de ellas son graduales y su magnitud depende de la concentración de los dos fármacos en interacción. Por todo ello, son difíciles de predecir y el ajuste de las dosis es complicado.
Dependencia de la dosis
En determinados casos, los valores de los parámetros farmacocinéticos varían según la dosis administrada, la concentración plasmática o el tiempo; por ejemplo, la disminución de la biodisponibilidad de la griseofulvina al aumentar la dosis, debido a su menor solubilidad en las secreciones del tracto gastrointestinales alto. Otro ejemplo es el aumento desproporcionado de la concentración en equilibrio estacionario de la difenilhidantoína al aumentar el intervalo de dosificación, porque las enzimas metabolizadoras de difenilhidantoína tienen una capacidad limitada para eliminar el fármaco y la velocidad de administración habitual se aproxima a la velocidad máxima de metabolización. Por último, la reducción de la concentración plasmática de carbamazepina cuando se administra de manera crónica porque este fármaco induce su propio metabolismo.
Otras causas de variabilidad cinética dependiente de la dosis son: saturación de la fijación a las proteínas plasmáticas y tisulares (fenilbutazona), secreción saturable en el riñón (dosis altas de penicilina) y metabolismo de primer paso hepático saturable (propranolol).
Los fármacos son casi siempre compuestos extraños al organismo. Como tales, no se están formando y eliminando continuamente al igual que sucede con las sustancias endógenas. Por tanto, los procesos de absorción, biodisponibilidad, distribución y eliminación tienen una importancia capital para determinar el inicio, la duración y la intensidad del efecto farmacológico.
ABSORCIÓN
Proceso de transporte del fármaco desde el lugar de administración hasta la circulación sistémica atravesando por lo menos una membrana celular.
La absorción de los fármacos viene determinada por sus propiedades físico-químicas, formulaciones y vías de administración.
Las formas en las que se presentan los medicamentos (p. ej., píldoras, comprimidos, cápsulas o soluciones) consisten en el fármaco y otros ingredientes. Los medicamentos se formulan para poder administrarlos por diversas vías (oral, bucal, sublingual, rectal, parenteral, tópica e inhalatoria). Un requisito esencial para que cualquier fármaco pueda absorberse es que sea capaz de disolverse. Los medicamentos sólidos (p. ej., los comprimidos) pueden disgregarse y desintegrarse, pero la absorción sólo ocurre cuando el principio activo se disuelve.
Transporte por las membranas celulares
Cuando los fármacos penetran en el organismo a través de la mayoría de las vías de administración (excepto la vía intravenosa o intrarterial), deben atravesar varias membranas celulares semipermeables antes de llegar a la circulación general.
Estas membranas actúan como barreras biológicas que, de modo selectivo, inhiben el paso de las moléculas del fármaco.
Las membranas celulares se componen fundamentalmente de una matriz lipídica bimolecular que contiene colesterol y fosfolípidos. Los lípidos proporcionan estabilidad a la membrana y determinan sus características de permeabilidad. En la matriz lipídica se encuentran embutidas macromoléculas proteicas globulares de volumen y composición variables. Algunas de estas proteínas de la membrana participan en el proceso de transporte y también pueden tener la función de receptores para la regulación celular.
Los fármacos atraviesan las barreras biológicas por
ð Difusión pasiva,
ð Difusión facilitada
ð transporte activo
ð Pinocitosis.
Difusión pasiva.
En este proceso, el transporte a través de la membrana celular depende del gradiente de concentración del soluto. La mayoría de las moléculas pasan a través de la membrana por difusión simple desde una zona con elevada concentración (p. ej., líquidos gastrointestinales ) hasta una zona de baja concentración (p. ej., la sangre). Puesto que las moléculas del fármaco son rápidamente transportadas a través de la circulación sistémica y se distribuyen enseguida en un gran volumen de líquidos corporales y tejidos, su concentración en el plasma es baja al principio, en comparación con la concentración en el lugar de administración; este amplio gradiente es la fuerza impulsora del proceso. La velocidad neta de difusión es directamente proporcional a este gradiente, pero depende también de la liposolubilidad, grado de ionización y tamaño molecular del fármaco y de la superficie de absorción.
Sin embargo, puesto que la membrana celular es de naturaleza lipídica, los fármacos liposolubles difunden con mayor rapidez que aquellos relativamente no liposolubles. Además, las moléculas pequeñas tienden a penetrar en las membranas con mayor rapidez que las de mayor volumen.
La mayoría de los fármacos son ácidos o bases orgánicas débiles que en medio acuoso están de forma ionizada y no ionizada. La importancia de este concepto está en que la fracción no ionizada suele ser liposoluble y difunde fácilmente a través de las membranas celulares. La forma ionizada no puede penetrar en las membranas tan fácilmente, debido a su baja liposolubilidad y elevada resistencia eléctrica. La resistencia eléctrica es resultante de la carga de la molécula y de los grupos con carga eléctrica de la superficie de la membrana. Por tanto, la penetración de un fármaco puede atribuirse principalmente a la fracción no ionizada.
La distribución de un fármaco ionizable a través de una membrana en el equilibrio viene determinada por el pKa de la sustancia que es la inversa de la constante de disociación (cuando el pH y el pK son iguales, las concentraciones de la forma ionizada y no ionizada del fármaco son iguales) y por el gradiente de pH, si existe.
En el caso de un ácido débil, cuanto mayor sea el pH, menor será el cociente entre la fracción no ionizada y la fracción ionizada. En el plasma (pH = 7,4), la proporción entre las formas no ionizada e ionizada de un ácido débil (p. ej., con un pKa de 4,4) es 1:1.000; en el jugo gástrico (pH = 1,4), la proporción se invierte, es decir, 1.000:1.
Por ejemplo:
Cuando el ácido débil, como la aspirina, se administra vía oral , el gradiente de concentración para la fracción no ionizada entre el estómago y el plasma tiende a aumentar, situación que favorece la difusión a través de la mucosa gástrica. Al alcanzar el equilibrio, las concentraciones de fármaco no ionizado en el estómago y en el plasma son idénticas porque sólo la forma no ionizada puede atravesar las membranas; la concentración de fármaco ionizado en plasma sería entonces aproximadamente 1.000 veces superior a la concentración de fármaco ionizado en la luz gástrica. En el caso de una base débil con un pKa de 4,4, la situación es la inversa.
Así, los fármacos que son ácidos débiles (p. ej., la aspirina), teóricamente deberían absorberse con mayor facilidad en un medio ácido (como la luz gástrica), que las bases débiles (p. ej., la quinidina). Sin embargo, independientemente del pH del fármaco, la mayor parte de la absorción tiene lugar en el intestino delgado por su extensión.
Difusión facilitada
Para ciertas moléculas (p.ej., glucosa), la velocidad de penetración es mayor a la esperada por su baja liposolubilidad. Se postula que existe un transportador que se combina de manera reversible con la molécula sustrato en la parte externa de la membrana celular y que el complejo transportador-sustrato difunde rápidamente a través de la membrana, liberando el sustrato en la superficie interna de la membrana. Este proceso de difusión mediado por un transportador se caracteriza por la selectividad y la saturabilidad. El transportador sólo acepta sustratos con una configuración molecular relativamente específica y el proceso está limitado por la disponibilidad de transportadores. Se trata de un mecanismo que no requiere energía, puesto que el sustrato no se transporta en contra de un gradiente de concentración.
Transporte activo
Además de la selectividad y de la capacidad de saturación, el transporte activo se caracteriza porque requiere gasto de energía por parte de la célula. Los sustratos pueden acumularse en el interior de la célula contra gradiente de concentración. Los procesos de transporte activo están limitados a los fármacos con similitud estructural con las sustancias endógenas. Estos fármacos suelen absorberse en lugares específicos del intestino delgado. Se han identificado procesos de transporte activo para diversos iones, vitaminas, azúcares y aminoácidos.
Pinocitosis
Consiste en el englobamiento y la captación de partículas o líquido por parte de una célula. La membrana celular se invagina, encierra a la partícula o al líquido y luego vuelve a fusionarse formando una vesícula que más tarde se desprende y emigra hacia el interior de la célula. Este mecanismo también requiere gasto de energía. Probablemente, la pinocitosis desempeña un papel menor en el transporte de fármacos, con la excepción de los fármacos que son proteínas.
Características de la administración oral
Puesto que la vía oral es el modo de administración más frecuente, la absorción suele referirse al transporte de los fármacos a través de las membranas de las células epiteliales del tracto gastrointestinales .
La absorción tras la administración oral depende de las diferencias del pH luminal a lo largo del tubo digestivo, de la superficie de absorción, de la perfusión tisular, de la presencia de flujo biliar y mucoso y de las membranas epiteliales.
Los ácidos se absorben más rápidamente en el intestino que en el estómago, en aparente contradicción con la hipótesis de que la forma no ionizada de un fármaco atraviesa con mayor facilidad las membranas. Sin embargo, esta discrepancia se debe a la enorme superficie del intestino delgado y a la mayor permeabilidad de sus membranas.
La mucosa oral posee un epitelio delgado y muy vascularizado que favorece la absorción, pero el contacto sucede por lo general por un tiempo demasiado corto, incluso para fármacos en solución, para que la absorción sea apreciable. En ocasiones, puede retenerse el fármaco durante más tiempo, para que la absorción sea más completa, situando el fármaco entre la encía y el carrillo (administración bucal) o colocándolo bajo la lengua ( sublingual ).
El estómago posee una superficie epitelial relativamente extensa, pero debido a la gruesa capa mucosa y a que el fármaco está en contacto relativamente poco tiempo, la absorción es limitada.
Dado que la absorción de prácticamente todos los fármacos es más rápida en el intestino delgado que en el estómago, la velocidad de vaciado gástrico es el paso limitante. Los alimentos, especialmente los grasos, enlentecen el vaciamiento gástrico (y la velocidad de absorción); este hecho explica por qué se recomienda tomar algunos fármacos con el estómago vacío cuando se desea que la acción comience rápidamente. La presencia de alimentos puede aumentar la absorción si el fármaco es poco soluble (p. ej., la griseofulvina); reducirla, si el fármaco se degrada en el estómago (p. ej., la penicilina G), o tener un efecto muy poco significativo o nulo. Además, los principios activos que alteran el vaciamiento gástrico (p. ej., los parasimpaticolíticos) también afectan la velocidad de absorción de otros fármacos.
El intestino delgado posee la mayor superficie para la absorción en el tracto gastrointestinales . En el duodeno el pH intraluminal oscila entre 4 y 5, pero se vuelve progresivamente más alcalino a lo largo del tubo digestivo (en la porción distal del íleon es cercano a 8). La flora gastrointestinales puede inactivar determinados fármacos, reduciendo así su absorción y su biodisponibilidad. La reducción del flujo sanguíneo (p. ej., en el shock) puede disminuir el gradiente de concentración a través de la mucosa intestinal y reducir la absorción por difusión pasiva. (La disminución del flujo sanguíneo periférico también altera la distribución y el metabolismo de los fármacos.)
La velocidad del tránsito intestinal puede influir en la absorción, especialmente en el caso de fármacos que se absorben por medio de transporte activo (p. ej., las vitaminas del complejo B), fármacos que se disuelven lentamente (p. ej., la griseofulvina) o los que son demasiado polares (poco liposolubles) para atravesar con facilidad las membranas (p. ej., muchos antibióticos). Para este tipo de fármacos, el tránsito debe ser muy lento para que la absorción sea completa.
Para las formas medicamentosas de liberación controlada, la absorción puede ocurrir inicialmente en el intestino grueso, especialmente cuando la liberación del principio activo de la forma medicamentosa dura más de 6 h, que es el tiempo de tránsito del intestino grueso.
Absorción de soluciones.
La absorción de los fármacos que se administran vía oral en forma de solución depende de si éstos son capaces de sobrevivir a los «encuentros» peligrosos con las numerosas secreciones gastrointestinales , los bajos pH y las enzimas potencialmente degradadoras. Por lo general, incluso si un fármaco es estable en el ambiente intestinal, una escasa fracción de él pasa al intestino grueso. Fármacos escasamente lipofílicos (de baja permeabilidad), como los aminoglucósidos, se absorben lentamente de la solución en el estómago y en el intestino delgado; para estos fármacos, la absorción por el intestino grueso se supone que será incluso más lenta, ya que la superficie de absorción es menor. En consecuencia, estos fármacos no son candidatos para formas de liberación controlada.
Absorción de formas sólidas.
La mayoría de los fármacos que se administran vía oral se presentan en forma de comprimidos o cápsulas, sobre todo por economía, estabilidad y aceptación por parte del paciente. Antes de absorberse se deben desintegrar y disolver. La desintegración aumenta considerablemente la superficie del fármaco que entra en contacto con los líquidos gastrointestinales y facilita su disolución y su absorción.
A menudo, durante el proceso de fabricación del medicamento se añaden los desintegrantes y otros excipientes (como disolventes, lubricantes, surfactantes, fijadores y dispersantes) para facilitar el proceso de desintegración.
Los surfactantes aumentan la velocidad de disolución del fármaco al incrementar su humectación, solubilidad y dispersabilidad. Entre los factores que modifican o retrasan la desintegración de las formas sólidas figuran la excesiva presión ejercida en la elaboración del comprimido y la aplicación de recubrimientos especiales para protegerlo de los procesos digestivos gastrointestinales . Los lubricantes hidrófobos (p. ej., el estearato de Mg) pueden fijar el principio activo y reducir su biodisponibilidad.
La velocidad de disolución determina la cantidad de fármaco disponible para la absorción. Cuando es más lenta que el proceso de absorción, la disolución constituye el paso limitante y puede manipularse por medio de cambios en la formulación del producto.
A menudo se emplea la reducción del tamaño de las partículas para aumentar la superficie del fármaco, lo cual resulta eficaz para aumentar la velocidad y la magnitud de la absorción gastrointestinales de un fármaco en el que estos parámetros están limitados por su lenta disolución. Entre los factores que afectan a la velocidad de disolución están si el fármaco está en forma de sal, en forma cristalina o en hidrato. Por ejemplo, las sales sódicas de los ácidos débiles (como barbitúricos y salicilatos) se disuelven más rápidamente que sus ácidos correspondientes, cualquiera que sea el pH del medio. Ciertos fármacos son polimorfos, pudiendo existir en forma amorfa o en varias formas cristalinas. El palmitato de cloranfenicol existe en dos formas, pero sólo una posee un grado suficiente de disolución para que su absorción sea de utilidad clínica. Cuando una o más moléculas de agua se combinan con un fármaco en forma cristalina se constituye un hidrato. La solubilidad del hidrato puede ser muy distinta de la que posee la forma no hidratada del compuesto; así, la ampicilina anhidra tiene mayor velocidad de disolución y de absorción que su trihidrato correspondiente.
Administración parenteral
La administración directa de un fármaco en el torrente circulatorio (habitualmente por vía intravenosa asegura la llegada de toda la dosis a la circulación general. Sin embargo, la administración del fármaco por una vía que requiera su paso a través de una o más membranas biológicas (inyección intramuscular o s.c.) para alcanzar la sangre no garantiza que se absorba totalmente. Para fármacos proteicos con PM >20.000 g/mol, el paso a través de las membranas de los capilares es tan lento que tras la administración intramuscular o subcutanea la mayor parte de la absorción se realiza a través del sistema linfático «por defecto». En estos casos, la velocidad de liberación a la circulación sistémica es lenta e incompleta, ya que hay un fenómeno de metabolismo de primer paso por las enzimas proteolíticas de los vasos linfáticos.
Como los capilares tienden a ser muy porosos, la perfusión (flujo sanguíneo por gramo de tejido) es el factor determinante de la velocidad de absorción en el caso de moléculas pequeñas. Por tanto, el lugar de inyección influye en la absorción del fármaco; así, la velocidad de absorción del diazepam inyectado por vía intramuscular en una zona con escaso flujo sanguíneo puede ser mucho más lenta que tras la administración de una dosis vía oral
Cuando se inyectan sales de ácidos o bases poco solubles por vía intramuscular , es posible que la absorción se retrase o sea errática. Por ejemplo, la forma parenteral de difenilhidantoína (fenitoína) es una solución de la sal sódica en propilenglicol al 40%, con un pH cercano a 12. Cuando se inyecta por vía intramuscular , el propilenglicol se absorbe y los líquidos hísticos, actuando como tampón, reducen el pH y producen un desplazamiento del equilibrio entre la forma ionizada y el ácido libre. Es entonces cuando el ácido libre, que es poco soluble, precipita. En consecuencia, la disolución y la absorción son muy lentas (entre 1 y 2 semanas).
Formas de liberación controlada
Las formas de liberación sostenida se han diseñado con el fin de reducir la frecuencia de administración y las fluctuaciones en las concentraciones plasmáticas y, de esta forma, conseguir un efecto farmacológico uniforme. Además, la menor frecuencia de administración es más cómoda para el paciente y puede mejorar el cumplimiento de la prescripción. En principio, los fármacos apropiados para este tipo de formas farmacéuticas son los que requieren una dosificación frecuente debido a su corta vida media de eliminación y a la breve duración de su efecto.
A menudo, las formas orales de liberación sostenida están diseñadas para mantener concentraciones terapéuticas del fármaco durante 12 h o más. La velocidad de absorción puede controlarse por distintas vías: recubriendo las partículas del fármaco con ceras u otras sustancias insolubles en agua, incluyendo al principio activo en una matriz de la que se libera lentamente a lo largo del tracto gastrointestinales o elaborando un complejo entre el fármaco y resinas de intercambio iónico.
Las formas tópicas de liberación sostenida se han diseñado para la presencia del fármaco durante períodos prolongados. Por ejemplo, la difusión de la clonidina a través de una membrana proporciona una liberación sostenida del fármaco durante 1 semana, y un polímero impregnado de nitroglicerina fijado a un vendaje adhesivo proporciona una liberación controlada durante 24 h. Los fármacos de liberación transdérmica deben poseer características adecuadas para la penetración cutánea y una potencia elevada, ya que la velocidad de penetración y el área de absorción son limitadas.
Se han formulado muchos preparados de administración parenteral no intravenosa, con el fin de proporcionar concentraciones plasmáticas sostenidas. En el caso de antibióticos, la inyección intramuscular de sales insolubles (p. ej., la penicilina G benzatina) consigue efectos farmacológicos clínicamente útiles durante largos períodos de tiempo. Otros fármacos están formulados como suspensiones o soluciones en vehículos no acuosos. Así, por ejemplo, la insulina puede inyectarse como suspensión cristalina para conseguir un efecto prolongado; la insulina amorfa, con una superficie de disolución mayor, posee un inicio del efecto rápido y una duración corta.
BIOEQUIVALENCIA Y BIODISPONIBILIDAD
Biodisponibilidad es la proporción de principio activo (fármaco o metabolito) que entra en la circulación general y que, por consiguiente, llega al lugar de acción, así como la velocidad con que ello sucede.
Mientras que las propiedades fisicoquímicas de un fármaco condicionan su potencial de absorción, las propiedades de la forma farmacéutica (de su diseño y de su manufactura) son determinantes principales de su biodisponibilidad. Las diferencias en la biodisponibilidad entre diferentes formulaciones de un mismo fármaco pueden ser clínicamente relevantes.
El concepto de equivalencia entre las formulaciones de un fármaco es importante a la hora de decidir el tratamiento más adecuado en cada situación.
El término equivalente químico (o farmacéutico) se refiere a los medicamentos que contienen el mismo principio activo en la misma cantidad y que cumplen los estándares oficiales; sin embargo, los ingredientes inactivos de los medicamentos pueden ser distintos.
La palabra bioequivalencia se aplica a los equivalentes químicos que, administrados a la misma persona siguiendo la misma pauta, alcanzan concentraciones similares en el plasma y en los tejidos. Los equivalentes terapéuticos designan dos medicamentos que, administrados a la misma persona y con la misma pauta, proporcionan esencialmente el mismo efecto terapéutico o tóxico. Se supone que los medicamentos bioequivalentes son terapéuticamente equivalentes.
Con frecuencia, algunos de los problemas terapéuticos (p. ej., toxicidad, pérdida de eficacia) que ocurren en el curso de tratamientos prolongados, cuando la enfermedad estaba siendo controlada con una formulación de un fármaco, son debidos al cambio por sustitutos no equivalentes (como en el caso de la digoxina o la difenilhidantoína).
En ocasiones es posible la equivalencia terapéutica aunque haya variaciones en la biodisponibilidad. Por ejemplo, el índice terapéutico (relación entre la dosis máxima tolerada y la dosis mínima eficaz) de la penicilina es tan amplio que diferencias moderadas en la concentración plasmática debidas a diferencias en la biodisponibilidad de las formulaciones de penicilina pueden no afectar la eficacia terapéutica o la seguridad del fármaco. Por el contrario, si se tratara de un fármaco con un índice terapéutico relativamente estrecho, las diferencias en la biodisponibilidad sí serían determinantes.
La biodisponibilidad también depende de otros factores, como los relacionados con la fisiología y las patologías -principal y asociadas- del paciente.
Este concepto es fundamental para explicarse por que las drogas no tienen siempre la misma magnitud de efecto
La velocidad a la que se absorbe un fármaco es un factor importante incluso cuando el fármaco se absorbe totalmente. Puede ocurrir que sea demasiado lenta para alcanzar una concentración plasmática terapéutica o tan rápida que se alcancen concentraciones tóxicas tras cada dosis.
Causas de baja biodisponibilidad
Cuando un fármaco se disuelve rápidamente de su formulación y atraviesa las membranas con facilidad, la absorción tiende a ser completa en la mayoría de las vías de administración. Éste no siempre es el caso de los fármacos administrados vía oral Antes de alcanzar la vena cava, un fármaco debe descender por el tracto gastrointestinales y atravesar la pared intestinal y el hígado, que son lugares donde habitualmente se metabolizan los fármacos ; por tanto, es posible que el fármaco se metabolice antes de llegar a la circulación general. Esta causa de baja biodisponibilidad se denomina metabolismo de primer paso. Muchos fármacos tienen una baja biodisponibilidad debido a que sufren un elevado metabolismo de primer paso. En muchos casos (p. ej.,isoproterenol, noradrenalina, testosterona), la extracción en esos tejidos es tan completa que la biodisponibilidad es prácticamente cero. Sin embargo, para fármacos con metabolitos activos, las consecuencias terapéuticas de este efecto de primer paso dependen de la contribución del fármaco o del metabolito a los efectos farmacológicos deseables o tóxicos.
La biodisponibilidad escasa es frecuente en formulaciones para administración oral de fármacos poco solubles en agua, que se absorben muy lentamente. Cuando la absorción es lenta o incompleta, los factores que pueden afectar la biodisponibilidad son más numerosos que cuando es rápida o completa. En el primer caso, puede esperarse una respuesta terapéutica mucho más variable.
La permanencia insuficiente del fármaco en el tracto gastrointestinales es una de las causas más comunes de biodisponibilidad escasa. Al ingerir un fármaco, éste no permanece en el tracto gastrointestinales más de 1-2 días y en el intestino delgado sólo se halla 2-4 horas. Si el fármaco no se disuelve con facilidad o si es incapaz de atravesar el epitelio intestinal (fármacos polares, muy ionizados), el tiempo en el que permanece en el lugar de absorción puede ser insuficiente. En estos casos, la biodisponibilidad no sólo es baja, sino que tiende a ser muy variable. Además, otros factores como edad, sexo, actividad, fenotipo genético, estrés, enfermedades (p. ej., aclorhidria, síndromes de mala absorción) o la cirugía gastrointestinales previa, pueden alterar e incluso aumentar todavía más las diferencias en la biodisponibilidad.
Las reacciones que compiten con la absorción pueden reducir la biodisponibilidad. Pueden ser la formación de complejos (p. ej., entre tetraciclina e iones metálicos polivalentes), la hidrólisis debida al ácido gástrico o a enzimas digestivas (p. ej., la hidrólisis de la penicilina y el palmitato de cloranfenicol), la conjugación en la pared intestinal (p. ej.,la sulfo-conjugación del isoproterenol), la adsorción por otros fármacos (p. ej., digoxina y colestiramina) y el metabolismo por la microflora intestinal.
Estimación de la biodisponibilidad
El análisis de la biodisponibilidad a partir de datos sobre la concentración plasmática respecto al tiempo suele requerir 3 medidas: la concentración plasmática máxima (pico) del fármaco, el tiempo en que aparece esta concentración máxima y el área bajo la curva concentración plasmática-tiempo
La concentración plasmática aumenta al hacerlo la velocidad y el grado de absorción; cuando la velocidad de eliminación del fármaco equivale a la velocidad de absorción, se alcanza el pico.
Las determinaciones de la biodisponibilidad basadas en la concentración plasmática máxima pueden ser erróneas, puesto que la eliminación empieza inmediatamente después de que el fármaco llega a la circulación. El tiempo en que se alcanza el pico plasmático depende de la velocidad de absorción; de hecho, es el índice más utilizado para medir este parámetro. Cuanto más lenta sea la absorción, más tarde se alcanzará el pico.
Sin embargo, a menudo el pico máximo no es un índice absolutamente definitivo, ya que se trata de un valor puntual, que depende de la frecuencia en la toma de muestras de sangre y, en caso de que las concentraciones cercanas al pico describan una curva relativamente plana, de la reproducibilidad de los resultados. Recordar que la concentración sanguínea de una droga muchas veces no es índice adecuado de la concentración en el sitio de acción.
Dosis única vs. dosis múltiples.
Aunque para medir la velocidad de absorción la administración única proporciona más datos que las administraciones múltiples, la biodisponibilidad puede medirse tras administraciones únicas o tras administraciones repetidas.
El estudio de los parámetros farmacocinéticos tras dosis múltiple posee algunas ventajas, como permitir representar de manera más exacta la situación clínica habitual. Normalmente se consiguen concentraciones plasmáticas superiores a las obtenidas con dosis única, lo que facilita su determinación.
Tras administrar dosis repetidas con intervalos regulares durante el período correspondiente a 4-5 vidas media beta o de de eliminación, las concentraciones plasmáticas deberían alcanzar el estado de equilibrio estacionario (la cantidad de fármaco absorbido se iguala con la cantidad de fármaco eliminado en cada intervalo de dosis).
En el caso de los fármacos que se excretan en forma inalterada (sin metabolizar) por la orina, puede estimarse la biodisponibilidad midiendo la cantidad total de fármaco excretado tras una administración única. Idealmente, debería recogerse la orina durante un período correspondiente a 7-10 vidas medias beta de eliminación con el fin de recuperar por completo el fármaco absorbido. En caso de múltiples dosis, la biodisponibilidad puede determinarse midiendo el fármaco inalterado en orina durante 24h en condiciones de equilibrio estacionario.
DISTRIBUCIÓN
Tras llegar a la circulación general, el fármaco pasa a los tejidos del organismo. Por lo común la distribución es desigual por las diferencias en la perfusión sanguínea, el grado de unión a los tejidos, las variaciones regionales del pH y la distinta permeabilidad de las membranas celulares.
La velocidad de penetración del fármaco en el tejido depende del flujo sanguíneo, de la masa de tejido y de la proporción del fármaco en sangre y en tejido.
En las zonas con una vascularización rica se alcanza el equilibrio de distribución (la velocidad de entrada y la velocidad de salida son iguales) entre el plasma y el tejido más rápidamente que en las zonas poco perfundidas, a no ser que la difusión a través de las membranas sea un paso limitante. Tras alcanzar el equilibrio de distribución, las concentraciones del fármaco (libre y unida a proteínas, v. más adelante) en los tejidos y en el líquido extracelular quedan reflejadas por la concentración plasmática. El metabolismo y la excreción tienen lugar simultáneamente con la distribución, lo que determina un proceso dinámico y complejo .
Volumen aparente de distribución
El volumen de líquido en el que parece distribuirse o diluirse el fármaco se denomina volumen aparente de distribución (el volumen corporal en que tendría que haberse disuelto el fármaco para alcanzar la misma concentración que en el plasma). Este parámetro informa sobre la concentración plasmática esperada para una dosis concreta y también sobre la dosis requerida del fármaco para obtener una concentración concreta. Sin embargo, el volumen aparente de distribución no proporciona datos sobre el patrón específico de distribución. Cada fármaco se distribuye en el organismo de un modo particular. Algunos tienden a dirigirse a los tejidos grasos, otros permanecen en el líquido extracelular y, por último, otros se fijan con avidez a tejidos específicos, como el hígado o el riñón.
Muchos fármacos ácidos (p. ej., la warfarina y el ácido salicílico) se fijan mucho a proteínas y, por tanto, tienen un volumen aparente de distribución pequeño. Muchos fármacos básicos (como la anfetamina y la meperidina) son captados con avidez por los tejidos y su volumen de distribución es mayor que el volumen de todo el organismo.
Fármaco unido ligado a proteínas
El grado de distribución de los fármacos en los tejidos depende de su unión a las proteínas plasmáticas y a diversos componentes tisulares.
Unión a proteínas plasmáticas
Los fármacos son transportados en la sangre en parte en solución (como fármaco libre, no unido) y en parte fijados a diversos componentes de la sangre (proteínas y células sanguíneas).
El principal determinante de la proporción entre el fármaco unido y el fármaco libre es la interacción reversible entre el fármaco y la proteína a la que se fija; esta interacción sigue la ley de acción de masas.
Muchas proteínas plasmáticas pueden interaccionar con los fármacos. Las más importantes son la albúmina, la glucoproteína ácida a1 y las lipoproteínas. Los fármacos de naturaleza ácida suelen fijarse a la albúmina, en tanto que los de tipo básico tienden a unirse a una de las dos últimas o a ambas
Puesto que sólo el fármaco libre puede sufrir una difusión pasiva hacia los tejidos y las zonas extravasculares en las que se ejerce el efecto farmacológico, la concentración de fármaco libre refleja mejor la concentración del fármaco en el lugar de acción y, por tanto, sus efectos.
La fracción libre (proporción de fármaco libre en relación a la concentración total) es un parámetro más útil que la fracción unida. La fijación a las proteínas plasmáticas influye en la distribución y en la relación aparente entre la actividad farmacológica y la concentración plasmática total del fármaco. A concentraciones elevadas de fármaco, la cantidad de fármaco unido se aproxima a un límite máximo, dependiendo del número de sitios de unión disponibles.
Por tanto, se dice que la fijación es saturable. La saturabilidad es la base de las interacciones por desplazamiento entre fármacos.
Fijación a los tejidos.
Los fármacos pueden unirse a muchas sustancias, además de a proteínas. Esta unión puede ser muy específica, como es el caso de la fijación de la cloroquina a los ácidos nucleicos. La unión tisular suele involucrar la asociación del fármaco con una macromolécula en un medio acuoso. Otro tipo de asociación que induce a pensar en una fijación tisular es la distribución del fármaco en la grasa corporal. Dado que el tejido adiposo está poco perfundido, el tiempo necesario para alcanzar el equilibrio en él es prolongado.
Reservorio de fármacos.
La acumulación en los tejidos o en los compartimientos corporales puede prolongar la permanencia de los fármacos en el plasma y sus acciones porque los tejidos sirven de depósito. A medida que la concentración plasmática disminuye, el fármaco almacenado se va liberando a la circulación. La localización del lugar de acción y las diferencias relativas en la distribución tisular también pueden ser importantes. El inductor anestésico tiopental, un tiobarbitúrico, es un ejemplo de fármaco cuyo almacenamiento en reservorios tisulares inicialmente acorta su efecto farmacológico, pero tras administraciones repetidas lo prolonga. El tiopental es un hipnótico muy liposoluble y se distribuye rápidamente en el cerebro tras la inyección intravenosa única. La concentración en el cerebro aumenta en 1 á 2 minutos, y posteriormente disminuye rápidamente en el cerebro y más lentamente la plasmática. La hipnosis finaliza a medida que el fármaco se redistribuye desde el cerebro al plasma y hacia los tejidos con perfusión más lenta, músculo y grasa. Sin embargo, si se determinan las concentraciones plasmáticas durante el tiempo suficiente, puede observarse una tercera fase de distribución que representa la liberación lenta del fármaco acumulado en el tejido adiposo. La administración continua de tiopental supone que grandes cantidades de fármaco se almacenan en el tejido graso, lo cual prolonga el efecto hipnótico.
Algunos fármacos se acumulan en las células en concentraciones superiores a las alcanzadas en el líquido extracelular. Esta acumulación suele implicar la fijación de fármacos a proteínas celulares, fosfolípidos o ácidos nucleicos. Los antipalúdicos como la cloroquina destacan por su notable fijación intracelular, de modo que pueden alcanzar concentraciones intracelulares en leucocitos y células hepáticas miles de veces superiores a las plasmáticas. El fármaco almacenado se encuentra en equilibrio con el plasmático y vuelve al plasma a medida que se va eliminando del organismo.
Barrera hematoencefálica
Los fármacos llegan al SNC por la circulación capilar y a través del LCR. Aunque el cerebro recibe una proporción importante del volumen minuto (aproximadamente 1/6), la distribución de los fármacos en el cerebro está restringida. Algunos fármacos liposolubles (como el tiopental) entran y ejercen sus efectos rápidamente, pero muchos otros -en particular los más hidrosolubles- penetran en el cerebro con mayor lentitud. Las células endoteliales de los capilares cerebrales están más estrechamente unidas entre sí que las de los demás lechos capilares del organismo; esto contribuye a la lenta penetración de las sustancias hidrosolubles. Otra barrera importante para los fármacos hidrosolubles son las células del tejido glial (los astrocitos) que forman una vaina pegada a la membrana basal del endotelio capilar.
El endotelio capilar y la vaina astrocítica constituyen la barrera hematoencefálica. Esta barrera es la que confiere las características diferenciales de permeabilidad entre estos tejidos y los del resto del organismo, en los que la barrera corresponde a la pared capilar y no a la célula parenquimatosa. Así, los compuestos polares son incapaces de penetrar en el cerebro, pero pueden acceder al líquido intersticial de la mayoría de los demás tejidos. El concepto de barrera hematoencefálica se definió tras la observación de que los colorantes polares podían penetrar en la mayoría de los tejidos, pero no en el SNC.
Los fármacos pueden pasar directamente al LCR ventricular a través del plexo coroideo, y tienen acceso al tejido cerebral por difusión pasiva desde el LCR. El plexo coroideo también es una zona de transporte activo de ácidos orgánicos (como la penicilina) desde el LCR a la sangre.
Los factores principales que determinan la velocidad de penetración en el LCR o en otras células son el grado de fijación a las proteínas, el grado de ionización y el cociente de partición lípido/agua del compuesto. La velocidad de penetración en el cerebro es lenta en los fármacos que se unen en gran proporción a proteínas. En el caso de ácidos y bases débiles ionizados, la penetración es tan lenta que se considera prácticamente inexistente.
En otros tejidos del organismo, la perfusión es el determinante principal de la velocidad de distribución, pero el SNC está tan bien perfundido que el factor más importante suele ser la permeabilidad. Sin embargo, en los tejidos poco perfundidos (p. ej., el músculo y el tejido adiposo), la distribución se prolonga notablemente, sobre todo si el tejido tiene mucha afinidad por el fármaco.
ELIMINACIÓN
Suma de procesos que conducen a la desaparición (por metabolimo y excreción) del fármaco del organismo.
METABOLISMO
El hígado es el órgano principal donde se produce el metabolismo de los fármacos (modificaciones químicas), pero no es el único. Algunos metabolitos tienen actividad farmacológica (v. tabla 298-2). Cuando la sustancia administrada es inactiva pero da lugar a un metabolito activo, el compuesto administrado se denomina profármaco, especialmente si ha sido diseñado para liberar eficazmente el principio activo.
Reacciones metabólicas
El metabolismo de los fármacos supone un amplio espectro de reacciones químicas:
ð oxidación,
ð reducción,
ð hidrólisis,
ð hidratación,
ð conjugación,
ð condensación
ð isomerización.
Las enzimas implicadas en estas reacciones están presentes en numerosos tejidos, pero, por lo general, se encuentran más concentradas en el hígado. Para muchos fármacos, el metabolismo se produce en dos fases. Las reacciones de fase I suponen la formación de un nuevo grupo funcional o una partición de la molécula (oxidación, reducción, hidrólisis); se trata de reacciones no sintéticas.
Las reacciones de fase II conllevan la conjugación con un compuesto endógeno (p. ej., ácido glucurónico, sulfato, glicina); se trata, pues, de reacciones sintéticas. Los metabolitos formados en las reacciones sintéticas son más polares y más fácilmente excretados por el riñón (en la orina) y por el hígado (en la bilis) que los formados en las reacciones no sintéticas. Algunos fármacos sufren procesos de metabolismo de ambos tipos. Pese a que se denominan fasesI y II, se trata, como puede verse, de una clasificación funcional, no secuencial, de las reacciones de metabolismo de fármacos.
Citocromo P-450.
El sistema enzimático más importante del metabolismo de fase I es el citocromo P-450, una superfamilia de enzimas microsomales que catalizan reacciones de oxidación de numerosos fármacos por su capacidad de transferencia de electrones.
Los electrones son aportados por la NADPH-citocromo P-450-reductasa, una flavoproteína que transfiere electrones del NADPH (la forma reducida del fosfato dinu-cleótido de nicotinamida-adenina) al citocromo P-450. Las enzimas del citocromo P-450 están agrupadas en 14 familias de genes de mamífero que comparten secuencias idénticas y 17subfamilias. Se denominan por un símbolo raíz (CYP), seguido de un numeral árabe para la familia, una letra para la subfamilia y otro número árabe para el gen específico.
Las enzimas de las subfamilias 1A, 2B, 2C, 2D y 3A son las más importantes del metabolismo en mamíferos. CYP1A2, CYP2C9, CYP2C19, CYP2D6 y CYP3A4 son los más importantes en el metabolismo humano. La especificidad de las enzimas permite explicar muchas interacciones entre fármacos. En la tabla 298-3 se presentan varios ejemplos de fármacos que interaccionan con enzimas específicas del complejo citocromo P-450 (v. también Interacciones farmacológicas, cap.301). Las diferencias genéticas entre pacientes pueden modificar la respuesta clínica.
Conjugación
La glucuronoconjugación es la reacción de fase II más común, y es la única que ocurre en el sistema enzimático microsomal hepático. Los glucurónidos se secretan por la bilis y se eliminan por la orina. El cloranfenicol, el meprobamato y la morfina son algunos ejemplos de fármacos metabolizados por esta vía.
La conjugación con aminoácidos, como la glutamina y la glicina, produce metabolitos (p. ej., ácido salicilúrico, de la conjugación de ácido salicílico y glicina) fácilmente excretables en la orina, pero que no suelen secretarse por la bilis. La acetilación es la vía metabólica principal de las sulfamidas. La hidralazina, la isoniazida y la procainamida también sufren acetilación. La sulfoconjugación es la reacción entre grupos fenol o alcohol y un sulfato inorgánico, que deriva en parte de aminoácidos que contienen azufre como la cisteína. Los ésteres de sulfato así obtenidos son polares y se excretan rápidamente en la orina. Algunos ejemplos de fármacos que forman sulfatos son: paracetamol, estradiol, metildopa, minoxidil y tiroxina. La metilación es la principal vía metabólica para inactivar algunas catecolaminas. La niacinamida y el tiouracilo también sufren procesos de metilación.
Modificaciones debidas a la edad
Los recién nacidos tienen un sistema enzimático microsomal hepático sólo parcialmente desarrollado y, en consecuencia, presentan algunas dificultades para metabolizar muchos fármacos (p. ej., hexobarbital, fenazetina, anfetamina y clorpromazina). La experiencia con el cloranfenicol en recién nacidos muestra claramente las graves consecuencias que se pueden derivar del enlentecimiento de la glucuronoconjugación. Dosis equivalentes en mg/kg de cloranfenicol, bien toleradas por pacientes mayores, pueden provocar una toxicidad grave en los recién nacidos (síndrome del niño gris), asociada a la presencia de niveles plasmáticos elevados de cloranfenicol durante largo tiempo.
A menudo, la capacidad metabólica también se encuentra disminuida en los pacientes ancianos; esta reducción varía en función del fármaco y no es tan grave como en los recién nacidos .
Reciclado
Una vez terminado el tratamiento para el que fueron prescritos, los medicamentos deben depositarse en un punto limpio, puesto que desecharlos indiscriminadamente junto con el resto de los residuos puede deteriorar gravemente el medio ambiente.
Iniciativa SIGRE (España)
Con el objetivo de cerrar correctamente el ciclo de vida de los medicamentos, la industria farmacéutica ha puesto en marcha SIGRE Medicamento y Medio Ambiente (Sistema Integrado de Gestión y Recogida de Envases), en colaboración con la farmacias y la distribución del sector, facilitando que los ciudadanos puedan desprenderse cómodamente, pero con todas la garantías sanitarias y medioambientales, de los restos de medicamentos y de sus envases a través del Punto SIGRE situado en las oficinas de farmacia.
SIGRE Medicamento y Medio Ambiente ofrece un tratamiento adecuado tanto a los residuos de los medicamentos como a sus envases, atendiendo a sus especiales características y evitando el posible daño medioambiental que estos causarían si entraran en contacto con otros residuos, con el agua de los ríos o los acuíferos o, en definitiva, con el entorno.
SIGRE cuenta con el control y apoyo de las Consejerías de Medio Ambiente, así como con la colaboración de las Consejerías Sanidad de las distintas Comunidades.
Polémica en la opinión pública
Las empresas con las que trabaja SIGRE-Danigal en la distribución y Sogama en la incineración de medicamentos- se ven envueltas en un escándalo hacia el año 2009 cuando salen a la actualidad informativa a causa de una filtración de información que acusa a Sogama a no realizar su cometido conforme a lo establecido en su acuerdo con SIGRE. En lugar de proceder a la incineración de medicamentos se confirma un enterramiento de los medicamentos (pastillas y demás material) en suelo gallego por la falta de los instalaciones (hornos) para llevarlo a cabo.
La noticia salió a la luz en La opinión de A Coruña a finales de enero de 2009, seguidamente al día siguiente El Mundo, ABC y Público también informaron sobre el tema. La Opinión de A Coruña realizó una labor de seguimiento respecto al fraude de la gestión de supuesta índole ecológica y se encargó de entrevistar al alcalde de la localidad (Cerceda) donde se acoge Sogama, liderada entonces por un gobierno de coalición Socialista y un partido local. No hace falta ser conocedor del contexto social y económico de Cerceda (A Coruña) para darnos cuenta rápidamente de que se trata de un pueblo pequeño con alrededor de 5.500 habitantes cuyo motor económico no está muy alejado de la planta de incineración Sogama. El alcalde, José García Liñares, responde con evasivas y no justifica los 32 millones de euros en ingresos en un pueblo eminentemente pequeño para tal volumen. La Opinión de A Coruña no quiso perder la oportunidad de comunicarse con el presidente de la Xunta Gallega y el consejero de medio ambiente, ambos respondieron con evasivas ante el tema remitiendo únicamente a las empresas responsables.
Ante esta situación de incertidumbre social y ecológica, la opinión pública silenciada en los medios de comunicación más generalistas emplea la única herramienta comunicativa que ha demostrado ser democrática y las opiniones y los debates tienen un lugar común: los foros y blogs en la red, donde ciudadanos de todas las partes de España establecen un diálogo y critican la mala gestión pública de una iniciativa en su origen ecológica y sin ánimo de lucro.
Normativa
España
La información que aparece en el etiquetado de los medicamentos, tanto del embalaje exterior como del acondicionamiento primario, así como los símbolos, siglas y leyendas que deben contener, se define en los anexos III y IV del Real Decreto 1345/2007, de 11 de octubre, por el que se regula el procedimiento de autorización, registro y condiciones de dispensación de los medicamentos de uso humano fabricados industrialmente.[32] [33] [34]
República Dominicana
Todo medicamento debe contar, antes de ser distribuido en este país, con un registro sanitario. Para obtener el registro sanitario en República Dominicana es necesario obtener permiso o autorización ante la Secretaría de Estado de Salud Pública del el ministerio de Salud. En República Dominicana, para iniciar la comercialización de alimentos, productos farmacéuticos, de uso doméstico y productos de cuidado personal. De acuerdo a lo anterior, el fabricante debe reunir ciertos requisitos, para obtener el Registro Sanitario, antes de proceder con el registro de productos, incluyendo:
Registrar ante el Ministerio de Salud una compañía autorizada de la distribución (compañía dominicana o una subsidiara de un fabricante incorporado bajo leyes dominicanas). En todo caso, para la obtención del Registro Sanitario, el fabricante puede decidir incorporar una compañía bajo leyes de República Dominicana para representarlo y distribuir los productos en el país y así evitar todas las obligaciones que establecen las leyes 173 sobre la comercialización y distribución de productos la cual otorga indemnizaciones altas a los distribuidores dominicanos en caso de rescisión (terminación) del contrato de distribución por parte del fabricante.
Véase también
- Adulteración
- Anexo:Medicamentos
- Cabás o Maletín médico
- Clasificación de Derivaciones Fármaco-terapéuticas
- Conservación de medicamentos
- Dispensación
- Dosis
Referencias
- ↑ Albarracín,A. et. al. 1.984 Historia del medicamento. Vol. I. Ed. Doyma S.A. Barcelona. 99 pp.
- ↑ Mª del Carmen Francés Causapé La Colección de medicamentos.
- ↑ Lastres, J.L. Director del Departamento de Farmacia y Tecnología Farmacéutica. Facultad de Farmacia - Universidad Complutense de Madrid. en la Página web de la Facultad de Farmacia y Bioquímica de la Universidad de Buenos Aires.
- ↑ a b c Domínguez-Gil,Alfonso. Catedrático de Tecnología Farmacéutica de la Universidad de Salamanca, en declaraciones a la revista Estar bien, edición digital, 8, octubre de 2008, nº 71
- ↑ Ley 29/2006 de Garantías y Uso Racional de los Medicamentos y Productos Sanitarios, en España.
- ↑ Medicamento. Diccionario de la Lengua Española (22ª ed.). Madrid: Real Academia Española; 2001.
- ↑ a b Cofepris.Glosario de Presentaciones Farmacéuticas 2006. Disponible en [1]. Consultado el 12 de octubre de 2008
- ↑ Hernandez Herrero, Gonzalo et al. MediPharm. Tratado de Medicina Farmacéutica. Editorial Médica Panamericana, 2010. ISBN 8498350107, 9788498350104
- ↑ Pharmaceuticals
- ↑ a b c d Dulanto, F. Dermatología médico-quirúrgica. Tomo II. Cap. 53. 1.341 pp. Ed. Anel s.a. Granada. 1.982 ISBN 84-85622-18-9
- ↑ [FN/2006/PO/034 en http://www.agemed.es/profHumana/farmacopea/docs/agua-alibour.pdf
- ↑ Sarkany, I T. St. John´s Hospital Dermatol. Soc., 59,241,1.973
- ↑ Korting, G.W. Manual de Dermatología Ed. Científico-Médica Barcelona 1986 28 pp ISBN 84-224-0813-9
- ↑ Leppard, B. Ashton, R. Tratamiento en Dermatología. Radcliffe Medical Press. Oxford. 1994. 5 pp ISBN 1-85775-003-9
- ↑ a b Galante, G.R. Soluciones para mucosas. Disponible en [2] Consultado el 12 de octubre de 2008
- ↑ Enrich, L. y Negre, S. Propuesta de clasificación de los apósitos estériles modernos. Cienc. Pharm. 1998 8(4): 153-171
- ↑ Hall, V.,Murillo, N.,Quesada,M. Apósitos hidrocoloides. Su papel en la curación de heridas. Centro de Información de Medicamentos. Marzo de 2001. disponible en [3]
- ↑ INSALUD. Guía práctica clínica: selección y utilización de medicamentos en las residencias geriátricas. 2000. España Disponible en [4]
- ↑ Serna, J. Vitales, M. López, M.C., Molina, A. Tomo II Capítulo 4: Dermatología en Farmacia Hospitalaria 868 pp, disponible en [5]
- ↑ a b Marcotegui Ros, F. Sistemas terapéuticos transdérmicos. Boletín de información farmacoterapéutica de Navarra. Vol 1 nº 3. 1993. Disponible en [6]
- ↑ «FDA approves scopolamine patch to prevent peri-operative nausea». Food and Drug Administration. Consultado el 12 febrero 2007.
- ↑ Rodriguez, R., Daza, P. y Rodriguez, M. F. Uso de buprenorfina transdérmica en el alivio del dolor por cáncer. Rev. Col. Anest. [online]. Oct./Dec. 2006, vol.34, no.4 [revisada el 10 de octubre de 2008], p.253-257. Disponible en [7]. ISSN 01203347.
- ↑ Clasificación Internacional de Patentes CIP 2007. Véase en [8]
- ↑ Memoria L. Novartis 2008. Disponible en [9]
- ↑ British Thoracic Society & Scottish Intercollegiate Guidelines Network (SIGN). British Guideline on the Management of Asthma. Guideline No. 63. Edinburgh:SIGN; 2004. (HTML, Full PDF, Summary PDF)
- ↑ Patente europea
- ↑ Formas farmacéuticas y vias de administración de fármacos. Departamento de Farmacología y Terapéutica. Facultad de Medicina. Universidad Autónoma de Madrid.
- ↑ El último avance en terapia gastrointestinal. El primer IBP desarrollado como isómero en Diariosalud.net
- ↑ Véase Conceptos básicos sobre Liposomas en [10]
- ↑ Richard Altschuler "DO MEDICATIONS REALLY EXPIRE?" Redflagsdaily, 2002. Republicación en Medscape
- ↑ Jasińska M et al "Stability studies of expired tablets of metoprolol tartrate and propranolol hydrochloride" Journal Acta Pol Pharm, 2009. PMID: 20050534
- ↑ Larrañaga Arregui B. Medicamentos de uso humano: etiquetado y prospecto, novedades del Real Decreto 1345/2007. Sendagaiak. 2008; 21(2):7-8.
- ↑ Real Decreto 1345/2007, de 11 de octubre, por el que se regula el procedimiento de autorización, registro y condiciones de dispensación de los medicamentos de uso humano fabricados industrialmente. BOE. 2007/11/07; nº 267.
- ↑ Real Decreto 1718/2010, de 17 de diciembre, sobre receta médica y órdenes de dispensación. BOE. 2011/01/20; (17):6306-29.
Enlaces externos
 Wikimedia Commons alberga contenido multimedia sobre Medicamento. Commons
Wikimedia Commons alberga contenido multimedia sobre Medicamento. Commons Wikcionario tiene definiciones para medicamento.Wikcionario
Wikcionario tiene definiciones para medicamento.Wikcionario- Agencia Española de Medicamentos y Productos Sanitarios
- Fichas técnicas de los medicamentos en España
- Diagnosia.com - Información de medicamentos
- Lista modelo de medicamentos esenciales de la OMS-2008
- Listado de Medicamentos Autorizados en España (uso humano)
- Organización Farmacéutica Colegial
- SIGRE Medicamento y Medio Ambiente
- Medicamentos
- Vademecum.com - Recursos sobre medicamentos
- Vademecum.es - Información de medicamentos








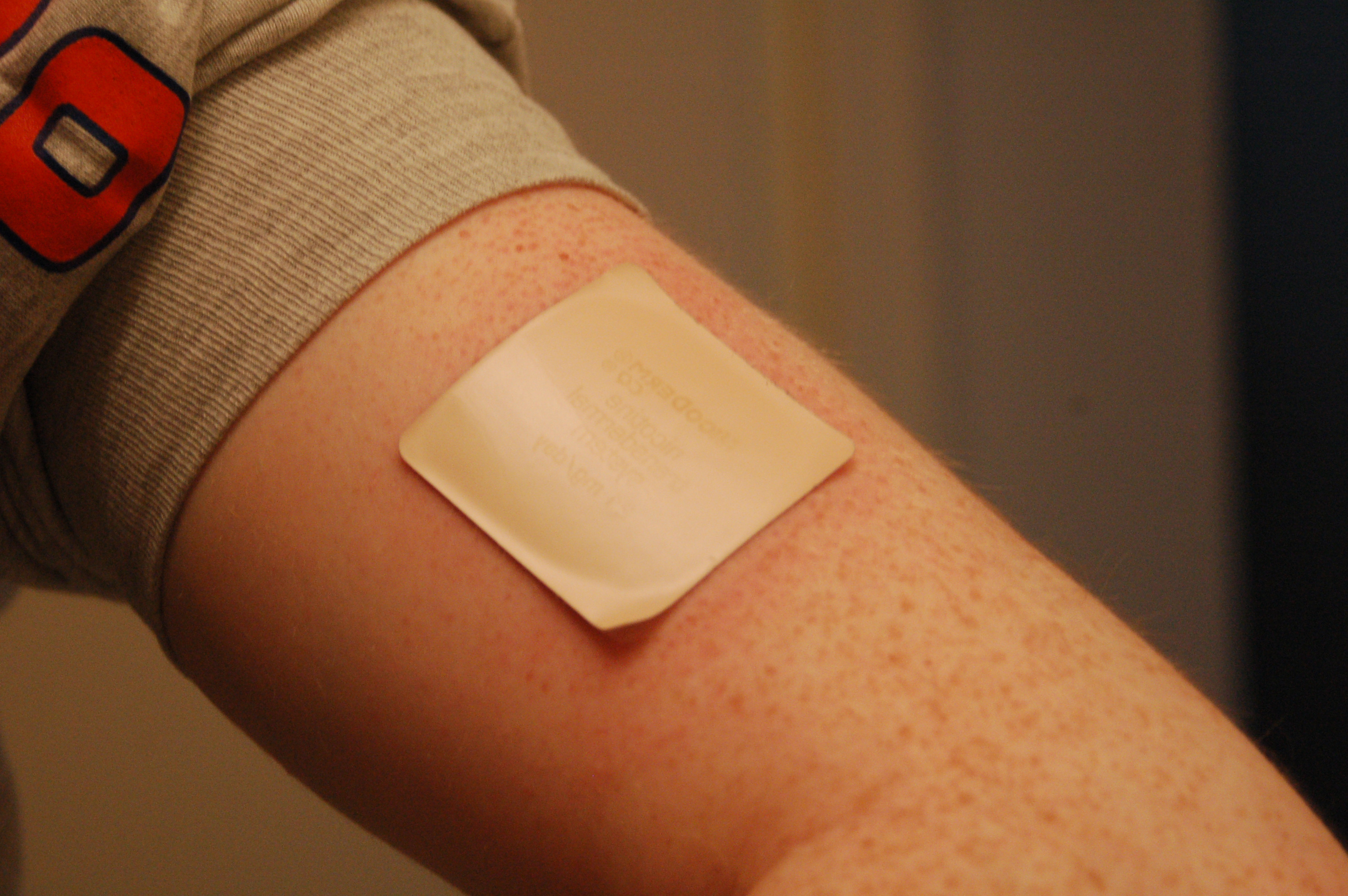

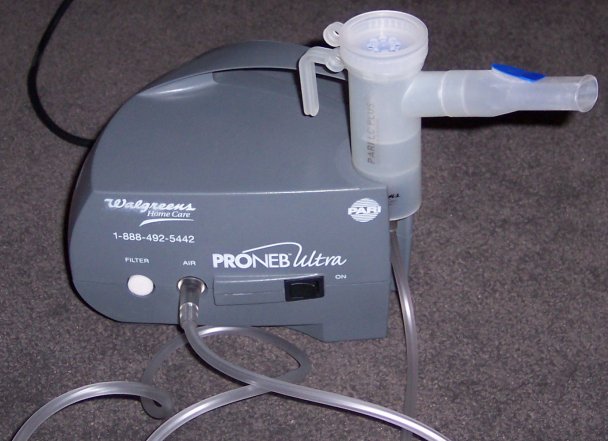
Wikimedia foundation. 2010.