- Secuenciación de ADN
-
La secuenciación de ADN es un conjunto de métodos y técnicas bioquímicas cuya finalidad es la determinación del orden de los nucleótidos (A, C, G y T) en un oligonucleótido de ADN. La secuencia de ADN constituye la información genética heredable del núcleo celular, los plásmidos, la mitocondria y cloroplastos (En plantas) que forman la base de los programas de desarrollo de los seres vivos. Así pues, determinar la secuencia de ADN es útil en el estudio de la investigación básica de los procesos biológicos fundamentales, así como en campos aplicados, como la investigación forense. El desarrollo de la secuenciación del ADN ha acelerado significativamente la investigación y los descubrimientos en biología. Las técnicas actuales permiten realizar esta secuenciación a gran velocidad, lo cual ha sido de gran importancia para proyectos de secuenciación a gran escala como el Proyecto Genoma Humano. Otros proyectos relacionados, en ocasiones fruto de la colaboración de científicos a escala mundial, han establecido la secuencia completa de ADN de muchos genomas de animales, plantas y microorganismos.

Los inicios
Durante treinta años la mayor parte de la secuenciación de ADN se llevó a cabo con el método de terminación de la cadena desarrollado por Frederick Sanger y colaboradores en 1975.[1] [2] Antes del desarrollo de métodos rápidos de secuenciación del ADN a principios de los 70 por Sanger en Inglaterra y Walter Gilbert y Allan Maxam en Harvard,[3] [4] se utilizaban varios métodos de laboratorio. Por ejemplo, en 1973[5] Gilbert y Maxam publicaron una secuencia de 24 pares de bases utilizando un método conocido como "de punto corrido" (wandering spot).
La secuenciación del ARN, que por razones técnicas es más sencilla de llevar a cabo que la del ADN, se desarrolló con anterioridad a la del ADN. El mayor hito en la secuenciación del ARN, que data de la era previa al ADN recombinante, es la secuencia del primer gen completo y del genoma completo del Bacteriófago MS2, identificado y publicado por Walter Fiers y colaboradores de la Universidad de Gante.[6] [7]
Secuenciación de Maxam-Gilbert
En 1976-1977, Allan Maxam y Walter Gilbert desarrollaron un método para secuenciar ADN basado en la modificación química del ADN y posterior escisión en bases específicas[8] Aunque Maxam y Gilber publicaron su secuenciación química dos años antes del trascendental artículo de Sanger y Coulson sobre su método de secuenciación "más-menos",[9] [10] la secuenciación de Maxam y Gilbert rápidamente se hizo más popular hasta que se pudo utilizar ADN directamente, mientras que el método inicial de Sanger requería que cada comienzo de lectura fuera clonado para producir un ADN de cadena simple. No obstante, con el desarrollo y mejora del método de terminación de la cadena (ver más adelante), la secuenciación de Maxam y Gilbert ha quedado en desuso debido a su complejidad técnica, el uso extensivo de productos químicos peligrosos y dificultades para escalarla. Además, a diferencia del método de terminación de la cadena, los reactivos que se usan en el método de Maxam y Gilbert no se pueden adaptar para utilizrse en un kit biológico estándar.
En resumen, el método requiere marcaje radiactivo en uno de los extremos y la purificación del fragmento de ADN que se desea secuenciar. El tratamiento químico genera rupturas en una pequeña proporción de uno o dos de los cuatro nucleótidos en cada una de las cuatro reacciones (G, A+G, C, C+T). De ese modo se genera una serie de fragmentos marcados a partir del final marcado radiactivamente hasta el primer lugar de "corte" en cada molécula. Los fragmentos posteriormente se separan por tamaño mediante electroforesis en gel, separando los productos de las cuatro reacciones en cuatro carreras distintas, pero una al lado de la otra. Para visualizar los fragmentos generados en cada reacción, se hace una autoradiografía del mismo, lo que proporciona una imagen de una serie de bandas oscuras correspondientes a los fragmentos marcados con el radioisótopo, a partir de las cuales se puede inferir la secuencia.
Conocido en ocasiones como "secuenciación química", este método se originó en el estudio de las interacciones entre ADN y proteínas (huella genética), estructura de los ácidos nucleicos y modificaciones epigenéticas del ADN, y es en estos campos donde aún tiene aplicaciones importantes.
Métodos de terminación de la cadena
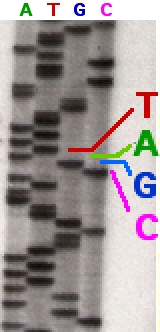
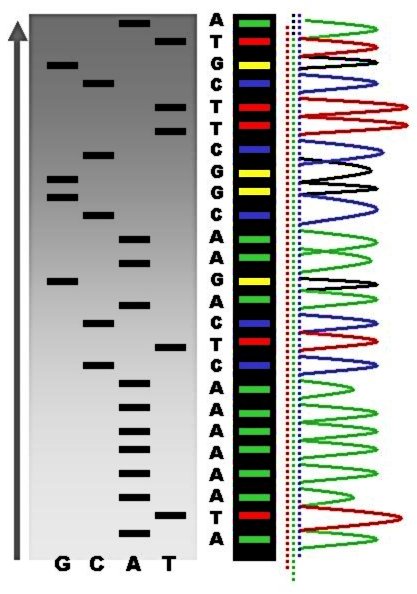
 Parte de un gel de secuenciación con marcaje radiactivo.
Parte de un gel de secuenciación con marcaje radiactivo.
Mientras que el método de secuenciación química de Maxam y Gilbert y el método más-menos de Sanger y Coulson eran órdenes de magnitud más rápidos que los métodos previos, el método de terminación de la cadena desarrollado por Sanger era incluso más eficiente y rápidamente se convirtió en el método de elección. La Técnica de Maxam-Gilbert requiere el uso de productos químicos altamente tóxicos y grandes cantidades de ADN marcado radiactivamente, mientras que el método de terminación de la cadena utiliza pocos reactivos tóxicos y cantidades menores de radiactividad. El principio clave del método de Sanger es el uso de didesoxinucleótidos trifosfato (ddNTPs) como terminadores de la cadena de ADN.
El método clásico de terminación de la cadena o método de Sanger necesita una hebra molde de ADN de cadena sencilla, un cebador de ADN, una ADN polimerasa con nucleótidos marcados radiactivamente o mediante fluorescencia y nucleótidos modificados que terminan la elongación de la cadena de ADN. La muestra de ADN se divide en cuatro reacciones de secuenciación separadas que contienen los cuatro desoxinucleótidos estándar (dATP, dGTP, dCTP and dTTP) y una ADN polimerasa. En cada reacción se añade solo uno de los cuatro didesoxinucleótidos (ddATP, ddGTP, ddCTP, o ddTTP). Estos didesoxinucleótidos terminan la elongación de la cadena al carecer un grupo 3'-OH que se necesita para la formación del enlace fosfodiéster entre dos nucleótidos durante la elongación de la cadena de ADN. La incorporación de un didesoxinucleótido en la cadena naciente de ADN termina su extensión, lo que produce varios fragmentos de ADN de longitud variable. Los didesoxinucleótidos se añaden a concentraciones lo suficientemente bajas como para que produzcan todas las posibilidades de fragmentos y al mismo tiempo sean suficientes para realizar la secuenciación.
Los fragmentos de ADN sintetizados y marcados de nuevo son desnaturalizados por calor y separados por tamaño (con una resolución de un solo nucleótido) mediante electroforesis en gel de poliacrilamida - urea. Cada una de las cuatro reacciones de síntesis se corre en carriles individuales (Carril A, T, G y C) y se visualizan las bandas de ADN mediante autoradiografía o luz ultravioleta, y la secuencia de ADN se puede leer directamente a partir de la placa de rayos X o de la imagen del gel. En la imagen de la derecha, la película de rayos-X se expuso directamente al gel de modo que las bandas oscuras corresponden a los fragmentos de ADN de diferentes longitudes. Una banda oscura en un carril indica un fragmento de ADN que es el resultado de una terminación de la cadena tras la incorporación de un didesoxinucleótido (ddATP, ddGTP, ddCTP, or ddTTP). El nucleótido terminal puede ser identificado de acuerdo al didesoxinucleótido que se añadió en la reacción que dio lugar a esa banda. Las posiciones relativas entre las cuatro calles se utilizan entonces para leer (de abajo a arriba) la secuencia de ADN como se indica.
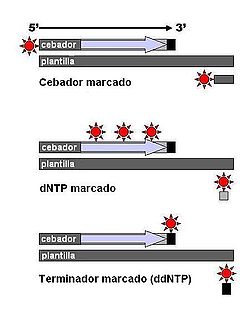 Los fragmentos de ADN se pueden marcar utilizando radiactividad o fluorescencia en el cebador (1), en la nueva cadena de ADN con dNTP marcado, o con un ddNTP marcado.
Los fragmentos de ADN se pueden marcar utilizando radiactividad o fluorescencia en el cebador (1), en la nueva cadena de ADN con dNTP marcado, o con un ddNTP marcado.Existen algunas variaciones técnicas del método de secuenciación de terminación de la cadena. En un método, los framentos de ADN son marcados con nucleótidos marcados con fósforo radiactivo. Como alternativa se puede utilizar un cebador marcado en el extremo 5' mediante un colorante fluorescente.
Se siguen necesitando cuatro reacciones, pero los fragmentos de ADN marcados con colorantes se pueden leer utilizando un sistema óptico, lo que facilita un análisis más rápido y económico y su automatización. Esta variante se conoce como "secuenciación mediante colorantes acoplados al cebador" (dye-primer sequencing). El último avance de L Hood y colaboradores[11] [12] desarrollando ddNTPs y cebadores con marcaje fluorescente señala el marco para una secuenciación de ADN automatizada y de alto rendimiento.
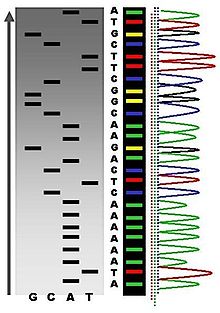 Escala de secuencia mediante secuenciación radiactiva comparada con máximos de fluorescencia.
Escala de secuencia mediante secuenciación radiactiva comparada con máximos de fluorescencia.Los diferentes métodos de terminación de la cadena han simplificado en gran medida la cantidad de trabajo y planificación necesaria para la secuenciación de ADN. Por ejemplo, el kit "Sequenase" de la casa USB Biochemicals, basado en el método de terminación de la cadena contiene la mayoría de los reactivos necesarios para la secuenciación, pre-divididos en alícuotas y listos para usar. Se pueden dar algunos problemas de secuenciación con el método de Sanger, como uniones no específicas del cebador al ADN, que afectan a la correcta interpretación de la secuencia de ADN. Además también puede afectar a la fidelidad de la secuencia obtenida estructuras secundarias internas de la cadena de ADN molde o ARN que pueda actuar de cebador al azar. Otros contaminantes que pueden afectar a la reacción son el ADN exógeno o inhibidores de la ADN polimerasa.
Secuenciación por terminador fluorescente
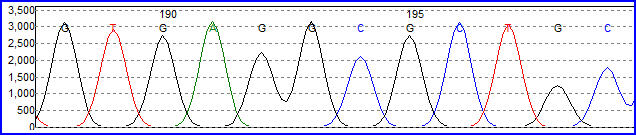
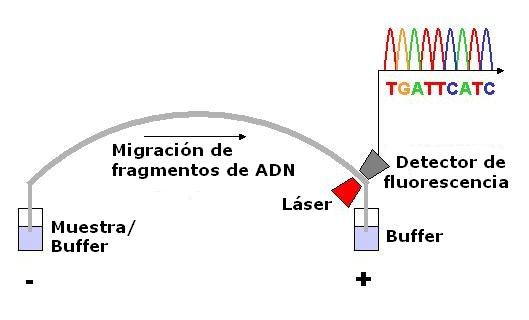
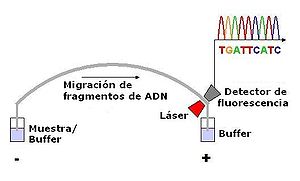 Electroforesis capilar.
Electroforesis capilar.Una alternativa al marcado del cebador es el marcado de los terminadores de la cadena, un método conocido como "secuenciación por terminador fluorescente". La mayor ventaja de este método es que la secuenciación se puede llevar a cabo en una sola reacción, en lugar de en cuatro reacciones como en el método del cebador marcado. En una secuenciación por terminador fluorescente se marcan cada uno de los cuatro didesoxinucleótidos que terminan la cadena con un colorante fluorescente diferente, con fluorescencias a diferentes longitudes de onda. Este método es atractivo por su gran capacidad y rapidez y actualmente es el método de referencia en la secuenciación automatizada con analizadores de secuencia controlados por computadora (ver más abajo). Entre sus limitaciones potenciales están los efectos de los terminadores fluorescentes en el fragmento de ADN, que produce alturas y formas de picos desiguales en los registros de secuencia de ADN del cromatograma tras la electroforesis capilar (ver ilustración de la derecha) Este problema se ha solventado en gran medida con la introducción de nuevos sistemas enzimáticos de polimerasas de ADN y colorantes que minimizan la variabilidad de la incorporación, así como métodos para eliminar los "pegotes de colorante" producidos por ciertas características químicas de los colorantes que pueden dar lugar a artefactos en los registros de secuencia de ADN. El método de secuenciado por terminador fluorescente junto con analizadores de secuencia de ADN de alto rendimiento se utiliza ahora para la inmensa mayoría de los proyectos de secuenciación, puesto que es más fácil de llevar a cabo y tiene un coste menor que los anteriores métodos de secuenciación.
Pirosecuenciación
La pirosecuenciación es un método de secuenciación de ADN en tiempo real basado en la liberación de los pirofosfatos (PPi) que tiene lugar en la reacción de polimerización del ADN a partir de sus dNTPs. Inicialmente, esta metodología se empleaba para monitorizar de forma continua la actividad de la ADN-polimerasa. Este método requiere de la preparación de una molécula monocatenaria de ADN a la cual se hibrida un pequeño cebador. A medida que la reacción transcurra, se irá sintetizando la cadena complementaria e iremos obteniendo una serie de picos de señal en el pirograma que nos permitirán determinar la secuencia. El proceso ocurre en ciclos sucesivos de tres pasos:
En el primero de ellos, se añade al medio de reacción uno de los 4 dNTPs el cual, si es el complementario a la base de la hebra molde que toca copiar, será procesado en la reacción de polimerización e incorporado a la cadena en extensión por la ADN-polimerasa liberando un PPi.
Dicho PPi es posteriormente convertido a ATP al reaccionar con una molécula de adenosina 5’-fosfosulfato por medio de la ATP-sulforilasa.
Finalmente, tiene lugar la emisión de radiación como consecuencia de la oxidación de la luciferina a oxiluciferina catalizada por la luciferasa de luciérnaga, consumiendo el ATP generado en la reacción anterior.
El número medio de fotones emitidos por cadena molde es proporcional al número de nucleótidos incorporados a la cadena, siendo esta relación lineal únicamente para un número bajo de incorporaciones. La radiación emitida es captada por una cámara acoplada a un sistema de cargas, el cual representa la señal en forma de pico en el pirograma. Si el dNTP que ha sido añadido al medio de reacción no es el complementario al que ocupa la posición que toca copiar, éste es degradado por una enzima llamada apirasa antes de que se añada el siguiente dNTP.
Así vemos que la información de la secuencia se representa en el pirograma en la que la altura de los picos refleja la cantidad de nucleótidos incorporada.
Secuenciación alelo-específica por bisulfito
La secuenciación por bisulfito es una variante de la secuenciación Sanger utilizada para el mapeo de metilaciones alelo-específicas en los sitios CpG. Se extrae el ADN a secuenciar y se fragmenta. A continuación se desnaturaliza y se incuba en presencia de bisulfito, el cual desaminará a las citosinas convirtiéndolas en
Automatización y preparación de las muestras
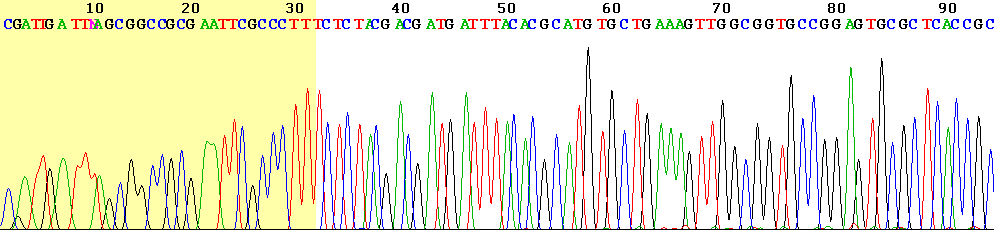
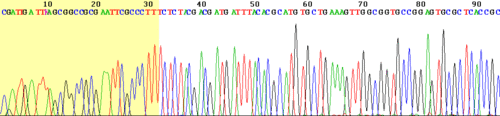 Vista del comienzo de un ejemplo de lectura con el terminador (fondo coloreado).
Vista del comienzo de un ejemplo de lectura con el terminador (fondo coloreado).Los instrumentos modernos automáticos de secuenciación del ADN (secuenciadores de ADN) pueden secuenciar más de 384 muestras marcadas por fluoresciencia de una sola vez y llevar a cabo 24 ciclos de secuenciación al día. No obstante, los secuenciadores automáticos de ADN llevan a cabo solamente separación del ADN basada en el tamaño (por electroforesis capilar), detección y registro de la coloración fluorescente, y los datos resultantes se dan como cromatogramas que registran los picos de fluorescencia. Se efectúan por separado las reacciones de secuenciación mediante una termocicladora, lavado y resuspensión en una solución tamponada antes de pasar las muestras al secuenciador. En el pasado los operadores tenían que arreglar los extremos terminales de baja calidad (ver imagen de la derecha) de cada secuencia manualmente para eliminar los errores de secuenciación. Sin embargo, hoy se puede realizar mediante software como "Fast Chromatogram Viewer" el arreglo automático de los extremos terminales en grandes cantidades.[13]
Estrategias de secuenciación a gran escala
Los procedimientos actuales solo pueden secuenciar directamente fragmentos relativamente cortos (de entre 300-1000 nucleótidos de longitud) en una sola reacción.[14] El principal obstáculo para secuenciar fragmentos de ADN de una longitud superior a este límite es la capacidad insuficiente de separación para resolver grandes framentos de ADN cuyo tamaño difiere en un sólo nucleótido. En cambio, las limitaciones impuestas por la incorporación de ddNTPs fueron resueltas en gran medida por Tabor, de la Hardvard Medical, Carl Fueller, de USB biochemicals, y colaboradores.[15]
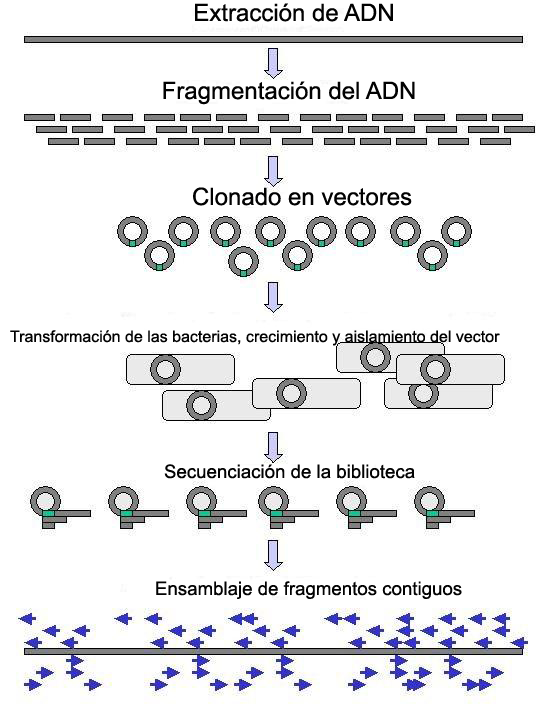
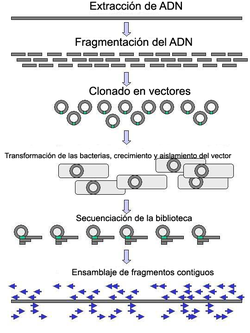 El ADN genómico se fragmenta en trozos al azar y se clonan en una biblioteca bacteriana. El ADN de los clones bacterianos individuales se secuencia y la secuencia se ensambla observando las regiones solapantes. (Click para ampliar)
El ADN genómico se fragmenta en trozos al azar y se clonan en una biblioteca bacteriana. El ADN de los clones bacterianos individuales se secuencia y la secuencia se ensambla observando las regiones solapantes. (Click para ampliar)La secuenciación a gran escala persigue la secuenciación de fragmentos muy grandes de ADN. Incluso los genomas bacterianos relativamente pequeños constan de miles de nucleótidos y sólo el Cromosoma 1 humano consta de 246 millones de bases. Así pues, algunos enfoques abordan el problema cortando (con enzimas de restricción) o cizallando (mediante fuerzas mecánicas) fragmentos grandes para obtener otros más pequeños. El ADN fragmentado se clona en un Vector de ADN, normalmente un cromosoma artificial bacteriano (BAC) y amplificado en Escherichia coli. El ADN amplificado se puede purificar entonces a partir de las células bacterianas (Una desventaja de los clones bacterianos para el secuenciado es que algunas secuencias de ADN pueden ser inherentemente inclonables en todas las líneas bacterianas disponibles debido al efecto deletéreo de la secuencia clonada en la bacteria hospedadora u otros efectos). Estos fragmentos cortos de ADN purificados a partir de colonias bacterianas individuales se secuencian completamente y se ensamblan computacionalmente en una secuencia larga y contigua identificando las secuencias que se solapan entre ellas (por secuenciación por fuerza bruta o "shotgun"). Este método no requiere información preexistente sobre la secuencia de ADN y a menudo se la conoce como secuenciación de novo. Los intervalos entre las secuencias ensambladas se pueden rellenar mediante paseos de cebadores, a menudo mediante pasos de sub-clonado (o secuenciación a base de transposones dependiendo del tamaño del resto de región que quede por secuenciar). Todas estas estrategias implican efectuar muchas lecturas menores del ADN por alguno de los métodos anteriores y posteriormente ensamblarlos en secuencias contiguas. Las diferentes estrategias tienen diferentes inconvenientes en cuanto a velocidad y exactitud. El método de secuenciación por fuerza bruta es el más práctico para secuenciar genomas grandes, pero su proceso de ensamblaje es complejo y potencialmente proclive al error -en particular en presencia de repetición de secuencias. Debido a esto, el ensamblaje del genoma humano no está literalmente completo — las secuencias repetitivas de los centrómeros, telómeros y otras partes del cromosoma quedan como huecos en el ensamblaje del genoma. A pesar de contar con solo el 93% del genoma ensamblado, el Proyecto Genoma Humano se declaró completado porque la definición de secuencia del genoma humano se limitó a la secuencia eucromática (completa al 99% en aquel momento), para excluir esas regiones repetitivas intratables.[16]
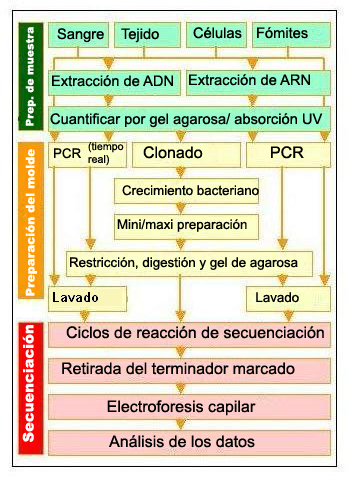
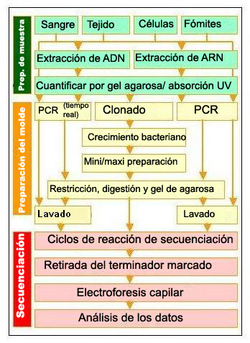 Pasos para la resecuenciación. Preparación de la muestra:Extracción del ácido nucleico. Preparación del molde:Amplificación y preparación de una pequeña región de la región objetivo. (Click para ampliar)
Pasos para la resecuenciación. Preparación de la muestra:Extracción del ácido nucleico. Preparación del molde:Amplificación y preparación de una pequeña región de la región objetivo. (Click para ampliar)El genoma humano tiene una longitud de unos 3000 millones de pares de bases;[17] si la longitud media de cada fragmento es de 500 bases, llevaría un mínimo de seis millones de fragmentos secuenciar el genoma humano (sin tener en cuenta el solapamiento, es decir si fuera posible hacerlo de una sola vez). Mantener el control de un número tan elevado de secuencias presenta desafíos significativos que solo se pueden abordar mediante el desarrollo y la coordinación de varios algoritmos de procedimiento y computación, tales como el desarrollo y mantenimiento eficientes de bases de datos.
Se utiliza la Resecuenciación o secuenciación marcada para determinar un cambio en la secuencia de ADN a partir de la secuencia "de referencia". A menudo se efectúa utilizando la PCR para amplificar la región de interés (se necesita una secuencia de ADN preexistente para diseñar los cebadores de ADN). La resecuenciación realiza tres pasos, la extracción del ADN o ARN del tejido biológico, la amplificación del ARN o ADN (habitualmente por PCR) y después la secuenciación. La secuencia resultante se compara con la de referencia o con una muestra normal para detectar mutaciones.
Nuevos métodos de secuenciación
Secuenciación de alto rendimiento
La elevada demanda de secuenciación de bajo coste ha dado lugar a las distintas tecnologías de secuenciación de alto rendimiento.[18] [19] Estos esfuerzos han sido financiados por instituciones públicas y privadas así como desarrolladas y comercializadas dentro de la empresa privada por las compañías de biotecnología. Se pretende que las tecnologías de secuenciación de alto rendimiento disminuyan los costes de secuenciación de las bibliotecas de ADN más allá de lo que se puede hacer con el método corriente del terminador marcado basado en la separación del ADN por electroforesis capilar. Muchos de los nuevos métodos de alto rendimiento usan métodos que paralelizan el proceso de secuenciación, produciendo miles o millones de secuencias a la vez.
- Amplificación clonal in vitro
Ya que los métodos de detección molecular frecuentemente no son lo suficientemente sensibles para la secuenciación de una sola molécula, la mayoría de los métodos utilizan un paso con clonación in vitro para generar muchas copias de cada molécula individual. Uno de los métodos es la PCR de emulsión, en la que se aislan las moléculas individuales de ADN junto con microesferas recubiertas con cebadores en burbujas acuosas dentro de una fase oleosa. Posteriormente una PCR recubre cada microesfera con copias clonales de la bliblioteca de moléculas aisadas y seguidamente se inmovilizan para ser más tarde secuenciadas. La PCR de emulsión se usa en los métodos publicados por Margulis y colaboradores (comercializado por 454 Life Sciences, adquirido por Roche), Shendure y Porreca et al. (conocido como "secuenciación polony ", —término formado por polimerasa "pol" y colonia "colony"), y la secuenciación SOLiD (desarrollada por Agencourt y adquirida por Applied Biosystems).[20] [21] [22] Otro método para la amplificación clonal in vitro es la "PCR de puente", en la que los fragmentos se amplifican a partir de los cebadores unidos a una superficie sólida, desarrollados y usados por Solexa (de la que ahora es propietaria la empresa Illumina). Estos métodos producen ambos muchas localizaciones físicamente aisladas que contienen cada una muchas copias de un solo fragmento. El método con una única molécula desarrolado por el laboratorio de Stephen Quake (y más tarde comercializado por Helicos) se salta este paso de amplificación, fijando directamente las moléculas de ADN a una superficie.[23]
- Secuenciación paralelizada
Una vez que las secuencias clonales de ADN se localizan físicamente en posiciones separadas de la superficie, se pueden utilizar varios métodos de secuenciación para determinar las secuencias de ADN de todas las localizaciones en paralelo. La "secuenciación por síntesis", como en la popular secuenciación electroforética con terminador marcado con colorante, usa el proceso de síntesis de ADN por ADN polimerasa para identificar las bases presentes en la molécula complementaria de ADN. Los métodos de terminador reversible (usados por Illumina y Helicos) utilizan versiones reversibles de terminadores marcados con colorante, añadiendo un nucleótido cada vez, y detectando la fluorescencia correspondiente a esa posición y removiendo posteriormente el grupo de bloqueo para permitir la polimerización de otro nucleótido. La Pirosecuenciación (utilizada por 454) también usa la polimerización del ADN para añadir nucleótidos, añadiendo cada vez un tipo diferente y después detectando y cuantificando el número de nucleótidos añadidos a una determinada localización a través de la luz emitida por la liberación de los pirofosfatos unidos a ellos.[20] [24]
La "secuenciación por ligación" es otro método enzimático de secuenciación que emplea una ADN ligasa en lugar de una polimerasa para identificar la secuencia objetivo.[25] [21] [22] Se usa en el método polony y en la tecnología SOLiD que ofrece Applied Biosystems. Este método utiliza un reservorio de todos los oligonucleótidos posibles de una longitud dada, marcados de acuerdo con la posición secuenciada. Los oligonucleótidos se templan y ligan; el ligamiento preferente de las ADN ligasas por su secuencia específica produce una señal correspondiente a la secuencia complementaria en esa posición concreta.
Otras tecnologías de secuenciación
Otros métodos de secuenciación por ADN podían tener ventajas en términos de eficiencia o exactitud. Al igual que la secuenciación por terminador marcado por tinción, están limitadas a la secuenciación de fragmentos únicos aislados. La "secuenciación por hibridación" es un método no enzimático que usa un chip de ADN. En este método, un único reservorio de ADN se marca mediante fluorescencia y se hibrida con un colección de secuencias conocidas. Si el ADN desconocido se hibrida fuertemente en un punto dado de entre las secuencias, haciéndole que "luzca", entonces se infiere que esa secuencia existe dentro de los ADN desconocidos que son secuenciados.[26] La Espectrometría de masas también se puede usar para secuenciar las moléculas de ADN; las reacciones convencionales de terminación de la cadena producen moléculas de ADN de diferentes longitudes y la longitud de esos fragmentos se determina entonces por las diferencias de masa entre ellas (en lugar de utilizar una separación por gel).[27]
Hay nuevas propuestas para la secuenciación de ADN que están en desarrollo, pero aún no han sido probadas. Entre estas están el marcaje de la ADN polimerasa,[28] la lectura de la secuencia a medida que la cadena de ADN pasa por nanoporos (secuenciación por nanoporos),[29] y técnicas basades en microscopías, como la microscopía de fuerza atómica o el microscopio electrónico que se usan para identificar las posiciones de los nucleótidos individuales dentro de largos fragmentos de ADN marcando los nucleótidos con elementos pesados (p.ej. halógenos) para la detección visual y su registro.[30] En octubre de 2006 el NIH publicó un boletín de noticias describiendo las nuevas técnicas de secuenciación y anunciando varias concesiones de becas.[31]
En octubre de 2006, la Fundación Premio X establecío el Premio Arconte X (Archon X Prize), que premia con 10 millones de dólares al "primer equipo que pueda construir un dispositivo y utilizarlo para secuenciar 100 genomas humanos en 10 días o menos, con una exactud no menor a un error por cada 100.000 bases secuenciadas, con secuencias que cubran correctamente al menos el 98% del genoma, y a un coste no mayor de 10.000 $ por genoma."[32]
Principales hitos en la secuenciación del ADN
- 1953 Descubrimiento de la estructura de la doble hélice de ADN.
- 1972 Desarrollo de la tecnología del ADN recombinante, que permite el aislamiento de fragmentos definidos de ADN; antes de este descubrimiento las únicas muestras accesibles para la secuenciación eran de bacteriófacos o virus de ADN.
- 1975 El primer genoma de ADN completamente secuenciado fue el del bacteriófago φX174
- 1977 Allan Maxam y Walter Gilbert publicaron el artículo "Secuenciación del ADN mediante degradación químicas.[33] Fred Sanger, independientemente, publica "Secuenciación del ADN mediante síntesis enzimática".
- 1980 Fred Sanger y Wally Gilbert reciben el Premio Nobel de química
- 1982 GenBank comienza su andadura como repositorio público de secuencias de ADN.
- Andre Marion y Sam Eletr de Hewlett Packard crean Applied Biosystems en Mayo, que acaba siendo hegemónica en la secuenciación automatizada.
- Akiyoshi Wada propone la secuenciación automatizada y recibe apoyo para la construcción de rebots con la ayuda de Hitachi.
- 1984 Los científicos del Medical Research Council descifran la secuencia completa de ADN del virus de Epstein-Barr, de una longitud de 170 Kbases.
- 1985 Kary Mullis y colaboradores desarrollan la reacción en cadena de la polimerasa, una técnica para replicar pequeños fragmentos de ADN.
- 1986 El laboratorio de Leroy E. Hood en el Instituto de Tecnología de California y Smith anuncian la primera máquina semiautomática de secuenciación de ADN.
- 1987 Applied Biosystems comercializa la primera máquina de secuenciación automatizada, el modelo ABI 370.
- Walter Gilbert abandona el panel sobre el genoma del Consejo Nacional de Investigación de los Estados Unidos para crear Genome Corp., cuyo objetivo es la secuenciación y comercialización de los datos.
- 1990 El Instituto Nacional de salud de los Estados Unidos comienza ensayos a gran escala de secuenciación de Mycoplasma capricolum, Escherichia coli, Caenorhabditis elegans, and Saccharomyces cerevisiae (a 75 centavos (US)/base).
- Lipman y Myers publican el algoritmo BLAST para la alineamiento de secuencias.
- Barry Karger (Enero[34] ), Lloyd Smith (Agosto[35] ), y Norman Dovichi (Septiembre[36] ) hacen una publicación sobre la electroforesis capilar.
- 1991 Craig Venter desarrolla la estrategia para encontrar genes que se expresan con ESTs (Expressed sequence tags).
- Uberbacher desarrolla GRAIL, un programa de predicción de genes.
- 1992 Craig Venter abandona el NIH para abrir el "The Institute for Genomic Research" (Instituto para la investigación genómica, El TIGR).
-
- El Trust Wellcome comienza su participación en el Proyecto Genoma Humano.
- Simon y al. desarrollan los BACs (cromosoma artificial bacteriano) para el clonado.
- Mapa físico del primer cromosoma publicado:
- Page y otros. - Cromosoma Y;[37]
- Cohen y otros. cromosoma 21.[38]
- Lander - mapa genético completo del ratón;[39]
- Weissenbach - mapa genético humano completo.[40]
- 1993 El trust Wellcome y MRC crean el Centro Sanger, cerca de Cambridge, Reino Unido.
- La base de datos GenBank se traslada de Los Álamos (DOE) al NCBI (NIH).
- 1995 Venter, Fraser y Smith publican la primera secuencia de un organismo de vida libre, Haemophilus influenzae (tamaño del genoma: 1.8 Mbase).
- 1996 Los socios internacionales del Proyecto Genoma Humano acuerdan publicar los datos de secuenciación en bases de datos públicas en 24 horas.
- Un consorcio internacional publica la secuencia del genoma de la levadura S. cerevisiae (tamaño del genoma 12.1 Mbases).
- Yoshihide Hayashizaki en el RIKEN completa el primer juego de cDNAs de longitud completa de ratón.
- ABI presenta un sistema de electroforesis capiar, el analizador de secuencias ABI310.
- 1997 Blattner, Plunkett y colaboradores. publican la secuencia de E. coli (Tamaño genómico 5 Mb)[42]
- 1998 Phil Green y Brent Ewing de la Universidad de Washington publican
“phred”para interpretar los datos de secuenciación(en uso desde 1995).[43]- Venter funda una nueva compañía, “Celera”; “Secuenciará el genoma humano en 3 años con un coste de $300m.”
- Applied Biosystems presenta la máquina de secuenciación capilar 3700.
- Wellcome dobla su financiación al Proyecto Genoma humano hasta $330 million para llegar a 1/3 de la secuencia.
- Objetivos del NIH y el DOE: Obtener un "borrador de trabajo" del genoma humano para 2001.
- Sulston, Waterston y otros finalizan la secuencia de C. elegans (tamaño del genoma de 97Mb).[44]
- 1999 El NIH cambia la fecha de finalización del borrador a la primavera de 2000.
- NIH lanza el proyecto de secuenciación del genoma del ratón.
- Primera secuencia del cromosoma humano 22 publicada.[45]
- 2000 Celera y colaboradores secuencian la mosca de la fruta Drosophila melanogaster (Tamaño del genoma 180Mb) - lo que supone la validación del método de Venter de secuenciación por fuerza bruta. El consorcio Proyecto Genoma Humano y Celera debaten sobre aspectos relacionados a la publicación de datos.
- El consorcio Proyecto Genoma Humano publica la secuencia del cromosoma 21.[46]
- HGP y Celera anuncian conjuntamente los borradores de trabajo de la secuencia del genoma humano y prometen una publicación conjunta.
- Las estimaciones del número de genes del genoma humano se sitúan entre 35.000 y 120.000. El consorcio internacional completa la primera secuencia de una planta, Arabidopsis thaliana (Tamaño del genoma 125 Mb).
- 2001 El Proyecto Genoma Humano publica el borrador de la secuencia del genoma humano en Nature el 15 de febrero.[47]
- Celera publica la secuencia del genoma humano.[48]
- 2005 420,000 secuencias VariantSEQr de cebadores de resecuenciación humanas publicadas en la nueva base de datos NCBI Probe.
- 2007 Por primera vez se secuencia un grupo de especies estrechamente relacionadas. Se secuencian 12 Drosofílidos (mosca de la fruta), haciendo despegar la era de la filogenómica.
- Craig Venter publica su genoma diploide completo: el primer genoma humano en ser completamente secuenciado.[49]
Véase también
- Análisis moleculares de ADN
- Secuenciación 454
- Proyecto Genoma Humano
- Medicina genómica
- Transistor de ADN de efecto de campo
Referencias
- ↑ Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975 Mayo 25;94(3):441–448
- ↑ Sanger F, Nicklen s, y Coulson AR, DNA sequencing with chain-terminating inhibitors, Proc Natl Acad Sci U S A. 1977 Diciembre; 74(12): 5463–5467
- ↑ Maxam AM, Gilbert W: A new method for sequencing DNA, Proc Natl Acad Sci U S A. 1977 Feb;74(2):560-4
- ↑ http://nobelprize.org/nobel_prizes/chemistry/laureates/1980/gilbert-lecture.pdf
- ↑ Walter Gilbert y Allan Maxam: The Nucleotide Sequence of the lac Operator, Proc Natl Acad Sci U S A. 1973 Diciembrer; 70(12 Pt 1-2): 3581–3584.
- ↑ Min Jou W, Haegeman G, Ysebaert M, Fiers W, Nucleotide sequence of the gene coding for the bacteriophage MS2 coat protein, Nature. 1972 Mayo 12;237(5350):82-8
- ↑ Complete nucleotide sequence of bacteriophage MS2 RNA: primary and secondary structure of the replicase gene. Nature. 1976 Apr 8;260(5551):500-7.
- ↑ Maxam AM y Gilbert W (1977). «A new method for sequencing DNA». Proc Natl Acad Sci U S A 2 (74). PMID 265521. http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=392330&blobtype=pdf.
- ↑ Sanger, F y Coulson, AR (1975) J. Mol. Biol. 94, 441-448
- ↑ Unknown
- ↑ Nature. 1986 Jun 12-18;321(6071):674-9. Fluorescence detection in automated DNA sequence analysis
- ↑ Nucleic Acids Res. 1985 Apr 11;13(7):2399-412. The synthesis of oligonucleotides containing an aliphatic amino group at the 5' terminus: synthesis of fluorescent DNA primers for use in DNA sequence analysis.
- ↑ Fast Chromatogram Viewer
- ↑ 3730xl DNA Analyzer
- ↑ a b «A novel thermostable polymerase for DNA sequencing.». Nature 376 (6543): pp. 796-797. 31-08-1995. PMID 7651542.
- ↑ International Human Genome Sequencing Consortium (2004). «Finishing the euchromatic sequence of the human genome.». Nature 431 (7011): pp. 931-45. PMID 15496913. paper available online
- ↑ Información del proyecto genoma humano
- ↑ Neil Hall (2007). «Advanced sequencing technologies and their wider impact in microbiology». The Journal of Experimental Biology 209: pp. 1518-1525.
- ↑ G.M. Church (2006). «Genomes for ALL». Scientific American 294 (1): pp. 47-54. PMID 16468433.
- ↑ a b M. Margulies, et al. (2005). «Genome sequencing in microfabricated high-density picolitre reactors». Nature 437: pp. 376-380.
- ↑ a b J. Shendure, G.J. Porreca, N.B. Reppas, X. Lin, J.Pe McCutcheon, A.M. Rosenbaum, M.D. Wang, K. Zhang, R.D. Mitra and G.M. Church. «Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome». Science 309 (5741): pp. 1728-1732.
- ↑ a b http://solid.appliedbiosystems.com/ - Applied Biosystems' SOLiD technology
- ↑ Braslavsky, I., Hebert, H., Kartalov, E. and Quake, S.R. (2003). «Sequence information can be obtained from single DNA molecules». Proceedings of the National Academy of Sciences of the United States of America 100: pp. 3960–3964.Texto completo disponible online
- ↑ M. Ronaghi, S. Karamohamed, B. Pettersson, M. Uhlen, and P. Nyren (1996). «Real-time DNA sequencing using detection of pyrophosphate release». Analytical Biochemistry 242: pp. 84=89.
- ↑ S. C. Macevicz, US Patent 5750341, filed 1995
- ↑ G.J. Hanna, V.A. Johnson, D.R. Kuritzkes, D.D. Richman, J. Martinez-Picado, L. Sutton, J.D. Hazelwood, R.T. D'Aquila (2000). «Comparison of sequencing by hybridization and cycle sequencing for genotyping of human immunodeficiency virus type 1 reverse transcriptase». Journal of Clinical Microbiology 38 (7): pp. 2715. PMID 10878069.
- ↑ J.R. Edwards, H.Ruparel, and J. Ju. «Mass-spectrometry DNA sequencing». Mutation Research 573 (1-2): pp. 3-12.
- ↑ VisiGen Biotechnologies Inc.
- ↑ Grupo de Harvard de Nanoporos
- ↑ Aplicación # 20060029957 de USPTO asignado a ZS genetics http://www.freepatentsonline.com/20060029957.html
- ↑ NHGRI pretende efectuar secuenciaciones de ADN más rápidas y baratas, NIH News Release, 4 October 2006
- ↑ "Panorama del Premio: Premio Arconte X de Genómica"
- ↑ A new method for sequencing DNA
- ↑ Karger, Barry L.; A. Guttman, A. S. Cohen, D. N. Heiger (15-01-1990). «Analytical and micropreparative ultrahigh resolution of oligonucleotides by polyacrylamide gel high-performance capillary electrophoresis». Analytical Chemistry 62 (2): pp. 137 - 141. doi 10.1021/ac00201a010.
- ↑ Smith, Lloyd M.; Luckey JA, Drossman H, Kostichka AJ, Mead DA, D'Cunha J, Norris TB (11-08-1990). «High speed DNA sequencing by capillary electrophoresis.». Nucleic Acids Research 18: pp. 4417-4421. PMID 2388826 doi 10.1093/nar/18.15.4417. http://nar.oxfordjournals.org/cgi/content/abstract/18/15/4417. Consultado el 2007-10-08.
- ↑ Dovichi, Norman J.; H.P. Swerdlow, S. Wu , H.R. Harke (07-09-1990). «Capillary gel electrophoresis for DNA sequencing: laser-induced fluorescence detection with the sheath flow cuvette». Journal of Chromatography 516: pp. 61-67. PMID 2286629.
- ↑ Page, DC; Foote S, Vollrath D, Hilton A (02-10-1992). «The human Y chromosome: overlapping DNA clones spanning the euchromatic region.». Science 258 (5079): pp. 60-66. PMID 1359640.
- ↑ Cohen, Daniel; Ilya Chumakov, Philippe Rigault, Sophie Guillou, Pierre Ougen, Alain Billaut, Ghislaine Guasconi, Patricia Gervy, Isabelle LeGall, Pascal Soularue, Laurent Grinas, Lydie Bougueleret, Christine Bellanné-Chantelot, Bruno Lacroix, Emmanuel Barillot, Philippe Gesnouin, Stuart Pook, Guy Vaysseix, Gerard Frelat, Annette Schmitz, Jean-Luc Sambucy, Assumpcio Bosch, Xavier Estivill, Jean Weissenbach, Alain Vignal, Harold Riethman, David Cox, David Patterson, Kathleen Gardiner, Masahira Hattori, Yoshiyuki Sakaki, Hitoshi Ichikawa, Misao Ohki, Denis Le Paslier, Roland Heilig, Stylianos Antonarakis (01-10-1992). «Continuum of overlapping clones spanning the entire human chromosome 21q». Nature 359 (6394): pp. 380-387. PMID 1406950 doi 10.1038/359380a0.
- ↑ Lander, E. S.; Dietrich W, Katz H, Lincoln SE, Shin HS, Friedman J, Dracopoli NC (1992-06). «A Genetic Map of the Mouse Suitable for Typing Intraspecific Crosses». Genetics 131: pp. 423-447. PMID 1353738. http://www.genetics.org/cgi/content/abstract/131/2/423.
- ↑ Weissenbach, Jean; Gyapay G, Dib C, Vignal A, Morissette J, Millasseau P, Vaysseix G, Lathrop M. (29-10-1992). «A second-generation linkage map of the human genome». Nature 359: pp. 794 - 801. PMID 1436057 doi 10.1038/359794a0.
- ↑ Mathies, R. A.; Ju J, Ruan C, Fuller CW, Glazer AN (09-05-1995). «Fluorescence Energy Transfer Dye-Labeled Primers for DNA Sequencing and Analysis». PNAS 92: pp. 4347-4351. PMID 7753809. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=7753809.
- ↑ «The Complete Genome Sequence of Escherichia coli K-12». Science 277 (5331): pp. 1453-1462. 05-09-1997. PMID 9278503 doi 10.1126/science.277.5331.1453.
- ↑ «Base-calling of automated sequencer traces using phred. II. Error probabilities.». Genome Reserarch 8 (3): pp. 186-94. 1998-03. PMID 9521922.
- ↑ «Genome Sequence of the Nematode C. elegans: A Platform for Investigating Biology». Science 282 (5396): pp. 2012-2018. 11-12-1998. PMID 9851916 doi 10.1126/science.282.5396.2012.
- ↑ «The DNA sequence of human chromosome 22». Nature 402 (6761): pp. 489-495. 02-12-1999. PMID 10591208.
- ↑ Hattori, M., A. Fujiyama, T. D. Taylor, H. Watanabe, T. Yada, H.-S. Park, A. Toyoda, K. Ishii, Y. Totoki, D.-K. Choi, E. Soeda, M. Ohki, T. Takagi, Y. Sakaki; S. Taudienk, K. Blechschmidtk, A. Polleyk, U. Menzelk, J. Delabar, K. Kumpfk, R. Lehmannk, D. Patterson, K. Reichwaldk, A. Rumpk, M. Schillhabelk, A. Schudyk, W. Zimmermannk, A. Rosenthalk; J. KudohI, K. ShibuyaI, K. KawasakiI, S. AsakawaI, A. ShintaniI, T. SasakiI, K. NagamineI, S. MitsuyamaI, S. E. Antonarakis, S. MinoshimaI, N. ShimizuI, G. Nordsiek, K. Hornischer, P. Brandt, M. Scharfe, O. SchoÈn, A. Desario, J. Reichelt, G. Kauer, H. Bloecker; J. Ramser, A. Beck, S. Klages, S. Hennig, L. Riesselmann, E. Dagand, T. Haaf, S. Wehrmeyer, K. Borzym, K. Gardiner, D. Nizetickk, F. Francis, H. Lehrach, R. Reinhardt, and M.-L. Yaspo, (2000). The DNA sequence of human chromosome 21. Nature 405: 311-319.
- ↑ «Initial sequencing and analysis of the human genome». Nature 409 (6822): pp. 860-921. 15-02-2001. PMID 11237011.
- ↑ Venter, J. C.; et al (16-02-2001). «The sequence of the human genome.». Science 291 (5507): pp. 1304-51. PMID 11181995 doi 10.1126/science.1058040.
- ↑ Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, et al. (2007) The Diploid Genome Sequence of an Individual Human. PLoS Biol 5(10): e254 doi:10.1371/journal.pbio.0050254
- Ansorge, W.J. (2009). «Next-generation DNA sequencing techniques.». New Biotechnology 25 (4).
- Diggle, M. A. (2004). «Pyrosequencing™». Molecular Biotechnology 28.
- França, L. T. C. (2002). «A review of DNA sequencing techniques.». Quarterly Reviews of Biophysics 35 (2).
- Hutchinson, C. A. (2007). «sequencing: bench to bedside and beyond.». Nucleic Acids Research 35 (18).
Enlaces externos
- Tecnología básica para el marcaje del ADN con átomos pesados para imágenes directas usando la microscopía electrónica de transmisión y la secuenciación de cadenas de más de 10.000 pares de bases por imagen capturada: Tecnología de marcaje con número Z alto.
- Plataforma de secuenciación de ADN Millegen
- Secuenciación de ADN: Animación de una reacción por terminador marcado por tinción
- Premio X de Archon - competición de 10 millones de $ para una tecnología de secuenciación rápida y barata.

Wikimedia foundation. 2010.