- Aminotransferasa
-
Las aminotransferasas (o transaminasas) son un conjunto de enzimas del grupo de las transferasas, pues transfieren grupos amino desde un metabolito a otro, generalmente aminoácidos. Estas enzimas son inducibles, porque su actividad puede aumentarse por la acción de diversas hormonas como la tiroxina o los glucocorticoides, su reacción es libremente reversible y su constante de equilibrio está cerca a la unidad.
Su nomenclatura se establece a partir del aminoácido des del cual transfieren el grupo amino. Los números EC 2.6 representan a las enzimas transferasas que transfieren grupos que contienen nitrógeno. Ver: Anexo:Números EC 2.6
Contenido
Mecanismos de la transaminación
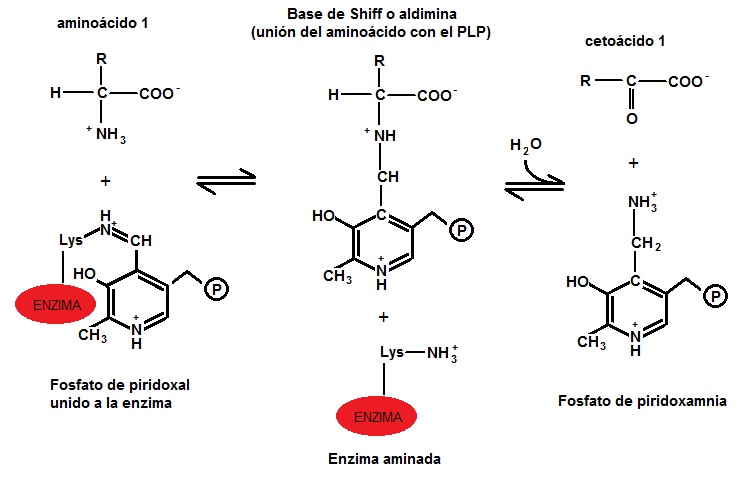
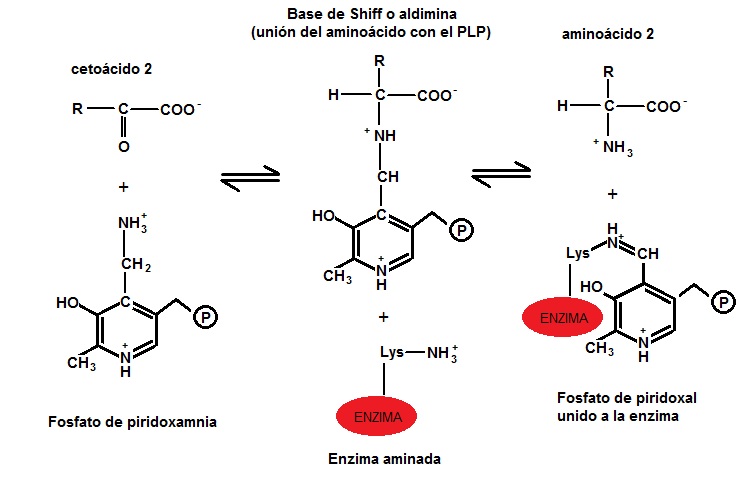
Las transaminasas necesitan de una coenzima llamada piridoxal fosfato (derivado de la piridoxina o vitamina B6) para ejercer su función; actúa como transportador del grupo amino entre los sustratos, alternando su estructura entre la forma aldehídica (piridoxal fosfato, PLP) y la forma aminada (piridoxamina-5-fosfato, PMP). El PLP contiene un anillo de piridina ligeramente básico y un hidroxilo que es ligeramente ácido, hecho que permite que sea muy estable porque es muy flexible. El grupo más importante del PLP es el aldehído. El piridoxal fosfato se une covalentemente al centro activo de las transaminasas a través del aminorante amino èpsilon de un residuo de lisina, y durante la reacción es transferido al aminoácido con formación de una base de Schiff, a partir de cuyo compuesto se producen las modificaciones químicas que conducen a la transaminación. Algunas aminotransferasas, sin embargo, utilizan el piruvato como cofactor.
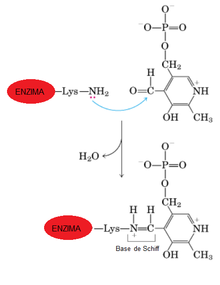 Unión de la coenzima PLP, que se modifica a Piridoxamina fosfato, al amino épsilon de un residuo de lisina de la enzima transaminasa
Unión de la coenzima PLP, que se modifica a Piridoxamina fosfato, al amino épsilon de un residuo de lisina de la enzima transaminasa
(Ver la imagen: Unión de la coenzima PLP, que se modifica a Piridoxamina fosfato, al amino épsilon de un residuo de lisina de la enzima transaminasa[1] )
Las transaminasas catalizan las reacciones de transaminación, importante sobre todo para la síntesis de aminoácidos no esenciales y la degradación de la mayoría de aminoácidos, que pierden su grupo amino por transaminación, excepto los aminoácidos lisina y treonina, donde esta reacción no es posible. Hay una aminotransferasa para cada aminoácido excepto para estos dos aminoácidos. Las principales aminotransferasas son las hepáticas como:
- La alanina aminotransferasa (ALT) o Glutamato-piruvato transaminasa (GPT), se localiza fundamentalmente a nivel citosólico en el hepatocito, por la que se la denomina unilocular.[2]
- La aspartato aminotransferasa (AST) o Glutamato-oxalacetato transaminasa (GOT), localizada sobretodo en la mitocondria y a nivel citoplasmático, por lo que se le llama enzima bilocular.[2] Ésta está presente, además del hígado, en otros órganos, como son, en orden de abundancia: el miocardio, el músculo esquelético, los riñones, el cerebro, el páncreas, el pulmón, los leucocitos y los eritrocitos.[3]
Estas aumentan en asociación a diversas enfermedades. En ocasiones, el tipo específico de aminotransferasa elevada sugiere el órgano afectado por su relativa abundancia en él.
En la transaminación participan normalmente, como donante y receptor, el glutamato y el α-cetoglutarato (α-KG), que participan en las diferentes reacciones catalizadas por las diferentes aminotransferasas. La transaminación consiste en transportar un grupo α-amino desde un α-aminoácido donador, al carbono ceto de un α-cetoácido receptor.[4] Este proceso ocurre en dos etapas[5] y es catalizado por las distintas aminotransferasas específicas de cada sustrato.
a) En la primera etapa un α-aminoácido que actuará como donador, transfiere el grupo α -amino a la enzima transaminasa, produciendo el correspondiente α-cetoácido y la enzima quedará aminada. (Ver imagen: Primera etapa de la transaminación)
b) En una segunda etapa, el grupo amino se transfiere al α-cetoácido aceptor (α-cetoglutarato, piruvato o oxalocetato) formando un nuevo aminoácido y regenerando la enzima. (Ver imagen: Segunda etapa de la transaminación)
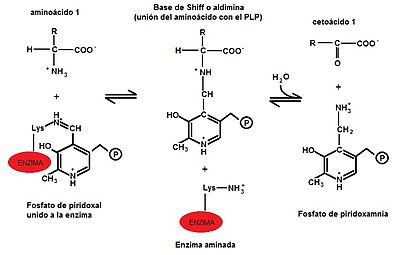
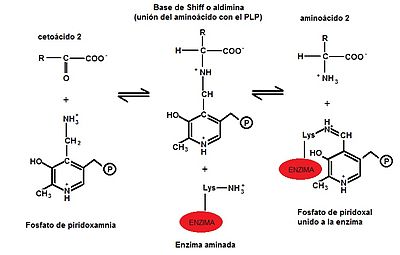
La reacción de la aminotransferasa ocurre vía mecanismo Ping Pong.[5]
Papel de las aminotransferasas en el metabolismo
Los humanos ingerimos nitrógeno a partir de aminoácidos de la dieta, proteínas y amoníaco que es fijado por las nitrogenasas bacterianas de las bacterias del intestino, el glutamato deshidrogenasa.[4] La glutamina sintasa convierten el amoníaco a glutamato y glutamina respectivamente, de los cuales las transaminasas transfieren sus grupos amino y amido a otros esqueletos de carbono por reacciones de transaminación y transamidación.
La reacción de transaminación tiene lugar en el citosol y las mitocondrias.[6] Las reacciones de transaminación son reversibles, así se podrán utilizar los α-cetoácidos para la síntesis de aminoácidos. Incluso si los α-cetoácidos que corresponden a los esqueletos de carbono de los aminoácidos esenciales son administrados por la dieta, podrán sintetizarse estos aminoácidos con una simple transaminación, catalizada por la aminotransferasa correspondiente.[4] El sentido de la reacción viene determinado por la concentraciones de productos y reactivos en el hígado porque, en éste, los metabólitos están próximos al equilibrio.
-
- La GOT cataliza la reacción hacia la formación de oxaloacetato.
- Aspartato + α-Cetoglutarato ⇔ Oxalacetato + Glutamato
-
- La GTP cataliza otra reacción, hacia la formación de piruvato.
- Alanina + α-Cetoglutarato ⇔ Piruvato + Glutamato
La GTP tiene una gran importancia en la catalización de reacciones que transfieren carbono y nitrógeno del músculo esquelético al hígado en forma de alanina. Primero, en el músculo esquelético, el piruvato actúa como receptor de un grupo amino y se transforma en alanina, esta será transportada a través del corriente sanguíneo hasta el hígado donde la alanina transferasa (ALT) transfiere el grupo amino al α-KG, regenerando así el piruvato que puede incorporarse en la gluconeogénesis como fuente de carbonos y la glucosa resultante podrá pasar de nuevo al músculo. Este proceso se conoce como el ciclo de la glucosa-alanina y permite la eliminación del nitrógeno del músculo esquelético en forma de urea, transformación que se dará gracias al Ciclo de la urea.
Síntesis de aminoácidos no esenciales
Los aminoácidos no esenciales son aquellos que pueden ser sintetizados por el cuerpo sin necesidad de ingerirlos y poder tener un correcto funcionamiento de todos los órganos, estos son: alanina, asparagina, aspartato, cisteina, glutamato, glutamina, glicina, prolina, serina y tirosina. La transaminación tendrá un papel importante en la síntesis de los aminoácidos no esenciales. La arginina, metionina y fenilalanina se pueden sintetizar en el cuerpo, pero a causa de que no podemos sintetizar la suficiente cantidad de estos aminoácidos para cubrir las funciones de nuestro metabolismo se consideran esenciales.
El glutamato se forma a partir de amoníaco y el α-cetoglutarato por una transaminación catalizada por la glutamato deshidrogenasa. El aspartato puede sintetizarse a partir de la asparagina o también se sintetiza con una transaminación catalizada por la aspartato aminotransferasa.[7]
La asparagina y la glutamina son sintetizadas por la asparagina sintetasa y glutamina sintetasa, respectivamente. La glutamina es producida por la fijación de nitrógeno a partir del glutamato y la asparagina se produce por una simple transaminación.[7]
Una unión de ATP y metionina forma S-adenosilmetionina, a la que se le añade un grupo SH para formar homcisteína, quién a su vez reaccionará con serina, y dará cistationina, que liberará un ion amonio y formará cisteína más α-cetobutirato.[7]
La tirosina se sintetiza a partir de fenilalanina mediante una reducción dependiente de NADH y es catalizada por la fenilalanina hidroxilasa que tiene como cofactor a la biopterina. La transaminación de fenilalanina da como producto el ácido fenilpirúvico, que se reduce a fenilacetato y fenilactato.[7]
La ornitina y la prolina derivan del glutamato. La ornitina se sintetiza a partir del glutamato cuando hay escasez de arginina en la dieta, la principal fuente de ornitina.[7]
La serina se sintetiza a partir del 3-fosfoglicerato, que se convierte en un cetoácido mediante una deshidrogenasa ligada a NADH. La serina puede ser precursor de glicina, mediante una transaminación catalizada por la serina hidroximetiltransferasa (SHMT1 si se trata de la enzima citosólica o SHMT2 si se trata de la enzima mitocondrial) en la que se transfiere el grupo hidroximetil de la serina al tetrahidrofolato (THF) obteniendo como productos glicina y N5,N10-metileno-THF. La glicina puede ser también precursora de la serina.[7] (vean imagen G) La glicina juega un papel importante en el anabolismo de nucleótidos de porina, glutatión, creatina, etc.
Las reacciones de transaminación son reversibles, en cambio las de transamidación necesitan ATP y son consideradas irreversibles.[7] El grupo α-amino es imprescindible para la síntesis de aminoácidos y deriva del amonio de los grupos aminos del L-glutamato. De estos se sintetizan glutamina, prolina y arginina. El ácido glutámico es la principal fuente de los grupos amino para la transaminación.
Degradación de aminoácidos
El exceso de nitrógeno potencialmente tóxico de los aminoácidos se elimina de la celula via transaminación, desaminación y formación de urea y los esqueletos de carbono pueden transformarse en carbohidratos con la incorporación a la gluconeogénesis o pueden conservarse como ácidos grasos con la incorporación a la vía de síntesis de ácidos grasos.[7]
Según los productos obtenidos en este proceso de degradación para eliminar el exceso de nitrógeno, los aminoácidos pueden clasificarse en glucogénicos, cetogénicos o glucogénicos y cetogénicos.[7] Los aminoácidos glucogénicos dan como producto de piruvato o intermediarios del ciclo del TCA o ciclo de Krebs, como son el α-cetoglutarato o el oxalocetato, precursores de la glucosa si se incorporan a la gluconeogénesis. Los aminoácidos únicamente cetogénicos son solos dos; la lisina y la leucina que dan como producto acetil-CoA o acetoacetil-CoA, de los quales no se puede producir glucosa. Los aminoácidos isoleucina, fenilalanina, treonina, triptófano y tirosina pueden dar productos precursores tanto de la glucosa como de los ácidos grasos, por eso se les clasifica como glucogénicos y cetogénicos.
Los aminoácidos no son utilizados como principal fuente de energía, aunque si no se necesitan para el recambio proteico, puesto que no se pueden almacenar, pueden utilizarse como tales.
La desaminación es el primer paso de todas las vías de degradación de aminoácidos que tiene lugar en la matriz mitocondrial.[6] Muchos aminoácidos son desaminados por transaminación. Las aminotransferasa remueven el grupo α-amino des del α-aminoácido donador hasta el carbono ceto de un α-cetoácido receptor (piruvato, oxalocetato o α-cetoglutarato). Si el aceptor del grupo amino es el cetoglutarato se producirá como nuevo aminoácido el glutamato.
Posteriormente se lleva a cabo una desaminación oxidativa, en la que la enzima ácido glutámico-deshidrogenasa elimina el grupo amino del ácido glutámico o glutamato. Esta reaccion requiere NAD+ i NADP+, regenera el cetoglutarato y se forma amoníaco que es tóxico para el cerebro.[8] El amoníaco se transportará hasta el hígado, donde tendrá lugar el Ciclo de la Urea que trasformará este compuesto en urea gracias a su unión con CO₂ para poder ser excretado. El cetoácido puede degradarse desaminandose por la vía del ácido cítrico o transformarse en glucosa por la vía de la gluconeogénesis, o en lípidos por la vía de la lipogénesis.
Nivel de Transaminasas en sangre
Los niveles de Transaminasas en sangre se utilizan como indicador para detectar posibles patologías en las funciones del hígado.
Tanto la AST y ALT están presentes en el suero en concentraciones inferiores a 30-40 Ul/l,[9] pero si el hígado está dañado, la permeabilidad de la membrana celular aumenta y estas enzimas son liberadas a la sangre en grandes cantidades, hecho que no siempre requiere la necrosis de los hepatocitos. De hecho, hay escasa correlación entre el daño celular hepático y el grado de elevación de las transaminasas. Prácticamente cualquier enfermedad hepática que comporte un daño necroinflamatorio puede ser la causa.[3]
Las enfermedades hepáticas -hepatitis viral, cirrosis-, el hígado graso, el consumo excesivo de alcohol, quistes o tumores en el hígado u obstrucción graves de la vía biliar pueden provocar un aumento notable de la transaminasa en sangre.
La elevación de transaminasas es un proceso muy inespecífico que puede ocurrir en casi todas las enfermedades hepáticas y en numerosas extrahepáticas.[3]
Las enfermedades hepáticas (hepatitis viral, cirrosis…) provocan un aumento notable de la transaminasa glutámico-pirúvico (ALT) en el plasma sanguíneo,[9] debido a su única localización en el hígado.[9] Otras enfermedades no hepáticas, como pueden ser aquellas relacionadas con procesos musculares (distrofias, polimiositis o traumatismos e un infarto agudo de miocardio) pueden ser la causa de un incremento más marcado de la transaminasa glutámico-oxalacético (AST), debido a su presencia, además del hígado, en otros órganos.[9]
Así pues, en la mayoría de tipos de enfermedad hepática, la actividad de la ALT es mayor que la de la AST.[9] La hepatitis alcohólica es una excepción a esta regla ya que el alcohol incrementa la actividad de la AST en el plasma, al contrario que otras formas de hepatitis; la mayoría de formas de daño hepático hacen disminuir la actividad hepatocitaria de ambas formas de la AST mientras que el alcohol sólo reduce la actividad citosólica. En los alcohólicos es común la deficiencia en piridoxina, que reduce la actividad de la AST y, finalmente, el alcohol induce la liberación de la AST mitocondrial a partir de células sin daño celular visible.[9]
Aún así, es prácticamente imposible que haya escasez de vitamina B6, ya que es una substancia que se encuentra en muchos alimentos: en carnes, yema de los huevos, grano integral, pescado, lácteos, frutas secas, etc.[10] Tampoco es común la ausencia de sustratos, ya que los aminoácidos no esenciales también se pueden ingerir por la dieta y prescindir de ser sintetizados a partir de los esenciales.
Pruebas de la función hepática
En medicina, el hecho de tener niveles más altos de lo normal de estas enzimas no indica, necesariamente, una enfermedad hepática establecida[9] y aún dándose el caso, existen varios tipos de daño hepático que puedan producir este efecto.[9] Así pues, la interpretación de los niveles altos de ALT y AST depende del cuadro clínico en general[3] (si el paciente presenta enfermedades sistemáticas asociadas, consumo de alcohol u otros fármacos, gravedad de los síntomas, si se acompaña de ictericia hepática…).[3] Por este motivo se realizan las llamadas pruebas de función hepática, que incluyen fosfatasa alcalina (FA), gamma glutamil transpeptidasa (GGT), albúmina, bilirrubina (total y directa) y estudio de coagulación. (Ver pruebas en la tabla)
 Pruebas de laboratorio que pueden identificar la causa de la hipertransaminemia
Pruebas de laboratorio que pueden identificar la causa de la hipertransaminemiaHipertransaminasemia aguda
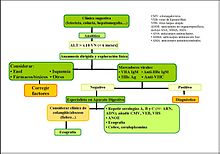 Hipertransaminasemia aguda
Hipertransaminasemia agudaUn caso de hipertransaminasemia por encima de diez veces su valor normal y de poca durada (inferior a 3-6 meses), conllevará a una necrosis hepática aguda o hepatitis aguda.[9] Cuando la ALT es superior a 1000 Ul/l la causa vendrá dada casi con toda seguridad por una hepatitis aguda viral (virus A, B y C) una hepatitis por fármacos o tóxicos o una hepatitis isquémica (fallo cardíaco agudo).[3] Las hepatitis víricas suponen la causa más frecuente de elevación de aminotransferasas, constituyendo más del 90% de los casos de hepatitis aguda, aunque deben investigarse otras causas.[9]
Con valores inferiores a 1000 Ul/l la hipertransaminasemia aguda puede ser debida al consumo de alcohol o ciertos fármacos, colangitis, Enfermedad de Wilson, hepatitis autoinmune, hepatitis por CMV, VEB y VHS, como también hepatitis por gérmenes infrecuentes (Brucella, fiebre Q, Leptospira, etc).
Procedimiento
Si un paciente presenta hepatits aguda, se considera primeramente que esta sea debida a un consumo de alcohol, una ingesta de medicamentos o un origen vírico, por lo que se utilizan los marcadores serológicos de infección viral por virus hepatotrópicos clásicos (anticuerpo anti-HA IgM, HBs Ag, anticuerpo anti-HBc IgM y anticuerpo anti-VHC). Si estas pruebas son negativas, se pasa a realizar otras para descartar causas más inusuales de hepatitis aguda, enfermedades hepáticas crónicas (sobre todo enfermedad de Wilson y hepatitis autoinmune) o patología biliar, por lo que se realiza una ecografía abdominal. (Ver pruebas en la tabla)
Se considerará preciso derivar al especialista del paciente cuando se detecte un fallo hepático agudo, en la presencia de un diagnóstico de hepatopatía de etiología poco frecuente, cuando haya la posibilidad de instaurar un tratamiento específico (por ejemplo: antivirales) o si la hepatopatía crónica se considera de gravedad y se ve necesario realizar un transplante. (Ver esquema: Hipertransaminasemia aguda)
Cuando se produce una curación de estas enfermedades se vuelve gradualmente a los valores normales de transaminasas en sangre.[11] Pero cuando el daño hepático se ha establecido de modo crónico o se ha producido una rotura notable de células hepáticas, con transformación cirrótica, la bajada de las transaminasas no indica curación sino que es señal de que ya no hay más células hepáticas que viertan estas enzimas en la sangre.[11]
Hipertransaminasemia prolongada
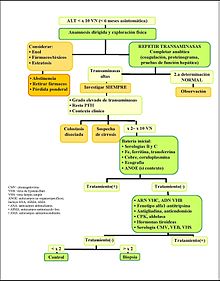 Hipertransaminasemia prolongada
Hipertransaminasemia prolongadaLa elevación de las transaminasas inferior a diez veces el valor normal con una duración superior a seis meses,[9] es la situación más frecuente en la práctica clínica. Se detectan muchos casos de manera accidental en pacientes asintomáticos (sin síndromes de enfermedad hepática o biliar) mediante analíticas rutinarias, donaciones sanguíneas,[9] estudios preoperatorios, etc. Entre el 1-4% de la población asintomática puede presentar elevación sérica de transaminasas.[3]
Las causas hepáticas pueden ser un abuso de fármacos, hepatitis crónica B, esteatosis hepática y esteatohepatitis no alcohólica, hepatitis autoinmune, hemocromatosis, Enfermedad de Wilson, déficit de Alfa 1-antitripsina, aunque las más frecuentes son el abuso de alcohol, esteatosis y la hepatitis por el virus C.[9] Las causas no hepáticas son la enfermedad celíaca, enfermedades hereditarias del músculo, enfermedades musculares adquiridas, ejercicio extenuante, patología tiroidea y suprarrenal y enfermedad inflamatoria intestinal crónica.[3]
Procedimiento
En la clínica, el primer paso es confirmar la persistencia pasadas 6-8 semanas de la elevación de las aminotransferasas del paciente (con el fin de confirmar una hipertransaminasemia prolongada), ya que muchos episodios de aumento de transaminasas se normalizan en un segundo control.[9] Si el paciente consume alcohol de manera habitual o es obeso será necesario que cambie sus hábitos durante este período y si éste consume algún tratamiento farmacológico deberá retirarlo siempre que sea posible.
Si las alteraciones analíticas persisten en el nuevo control analítico, es necesario iniciar una investigación sistematizada de las distintas causas hepáticas. Se realizan pruebas varias que incluyen la bilirrubina, GGT, FA (enzimas hepáticas que pueden ser útiles a la hora de orientar la etiología del proceso, por ejemplo hacia una patología colostática),[9] glucemia, colesterol y triglicéridos, hemograma, tiempo de protrombina, proteinograma y determinación de inmunoglobulinas, marcadores de infección viral crónica, hierro y ferritina y transferrina plasmáticos, como también la realización de una ecografía abdominal. (Ver esquema: Hipertransaminasemia prolongada)
Si aún así todavía no se dispone de un diagnóstico se tendrá en cuenta que enfermedades no hepáticas puedan ser la causa. Si tampoco se detecta la causa, será necesario un seguimiento clínico y analítico.
Cuando se considere la posibilidad de un tratamiento específico (por ejemplo: antivirales), o si la hepatopatía crónica se considera de gravedad y se ve necesario realizar un trasplante se considerará derivar el paciente al especialista.
Notas y referencias
- ↑ Tejedor, Cristina (2010). «Reacciones generales del metabolismo de los aminoácidos» (en español). Consultado el 29 de noviembre de 2010.
- ↑ a b Brandan, Nora (2008). «Enzimas» (en español) (pdf ubicación= Universidad Nacional del Nordeste Facultad de Medicina Càtedra de Boquímica). Consultado el 1 de diciembre de 2010.
- ↑ a b c d e f g h Díaz Otero, Arantxa. «Hipertransaminasemia» (en español).
- ↑ a b c King, Michael (28 de noviembre de 2010). «Metabolismo del Nitrógeno» (en español). Consultado el 1 de diciembre de 2010.
- ↑ a b Vázquez Contreras, Edgar (10 de octubre de 2003). «Transaminación de los aminoácidos» (en español). Consultado el 24 de noviembre de 2010.
- ↑ a b «Metabolismo Interno. Metabolismo de las proteinas» (en español) (2005). Consultado el 1 de diciembre de 2010.
- ↑ a b c d e f g h i W. King, Ph.D, Michael (12 de noviembre de 2010). «Metabolismo de los aminoácidos» (en español). Consultado el 17 de noviembre de 2010.
- ↑ Vázquez Contreras, Edgar (10 de octubre de 2003). «Desaminación» (en español). Consultado el 24 de noviembrede 2010.
- ↑ a b c d e f g h i j k l m n ñ Cuadrado, A.; Crespo, J. (2004). «Hipertransaminasemia en pacientes con negatividad de marcadores virales». Revista Española de Enfermedades Digestivas 96 (7). ISSN 1130-0108.
- ↑ Licata, Marcela. «Vitamina B6 - Piridoxina» (en español). Consultado el 1 de noviembre de 2010.
- ↑ a b «Transaminasas» (en español). Consultado el 10 de noviembre de 2010.
Bibliografía
Strayer, Lubert; Tymoczko, John; Jeremy, Berg (2004) (en Español). Bioquímica (quinta edición). Barcelona: Reverté.
Lehninger, Albert (en Inglés). Biochemestry (sexta edición). California: NW Freeman.
Peña, Antonio (1995) (en Español). ¿Cómo funciona una célula? Fisiología Celular (primeria edición). México. ISBN 968-16- 4365-8. http://www.vi.cl/foro/topic/7260-zcomo-funciona-una-celula-fisiologia-celular-libro-completo/. Consultado el 13 de diciembre de 2010.
Cackyne, Susan (1995) (en Español). Química Clínica. México: Interamericana.
Enlaces externos
- Transaminasas.com Artículos sobre las transaminasas.
Categorías:- Enzimas
- Hepatología
- Transferasas
Wikimedia foundation. 2010.