- Hemocromatosis
-
Hemocromatosis 
Preparación teñida con azul de Prusia para revelar depósitos de hierro; muestra un patrón típico de acumulación de hierro de la hemocromatosis genética en heterocigosis (véase coloración azulada al ampliar la imagen).Clasificación y recursos externos CIE-10 E83.1 CIE-9 275.0 OMIM 235200 MedlinePlus Información de salud en la enciclopedia MedlinePlus Sinónimos - Enfermedad de von Recklinghausen-Applebaum (forma hereditaria)
* Diabetes bronceada
 Aviso médico
Aviso médico La hemocromatosis (del griego αἷμα, haima: sangre y χρώμα, chróma: color)[1] es una enfermedad hereditaria que afecta al metabolismo del hierro, provocando un acúmulo excesivo e incorrecto de este metal en los órganos y sistemas del organismo. No se debe confundir con la hemosiderosis,[2] afección caracterizada por el exceso de hemosiderina en los tejidos, que no llega a producir daño orgánico. Cuando el depósito de hierro es tal que ocasiona perjuicio a los órganos en los que se acumula (especialmente el hígado), se habla de hemocromatosis.
En concentraciones fisiológicas, el hierro es un elemento vital para el organismo gracias a su capacidad de recibir y ceder electrones. Sin embargo, cuando se encuentra en grandes cantidades pierde esta función y genera radicales libres,[3] causantes del daño orgánico presente en la enfermedad.
Contenido
Historia
 Armand Trousseau, descubrió la hemocromatosis.
Armand Trousseau, descubrió la hemocromatosis.
La enfermedad fue descubierta por Armand Trousseau en 1865,[4] quien describió un síndrome clínico representado por diabetes, hiperpigmentación cutánea y cirrosis hepática. A esta asociación, en 1871, Troisier la llamó diabetes bronceada.
Fue en 1889 cuando se acuñó el nombre de hemocromatosis para designar el síndrome clínico de Trousseau. Fue von Recklinghausen quien le dio dicha nominación tras descubrir que el hierro era el metal acumulado en los tejidos. También fue quien comenzó a sospechar de su carácter hereditario.
En 1935, Joseph H. Sheldon afianzó los estudios de Recklinghausen y confirmó que se trataba de una enfermedad genética que alteraba el metabolismo del hierro.
Sin embargo, no fue hasta 1975 cuando Simon y colaboradores lograron demostrar la patogenia genética de la enfermedad mediante el descubrimiento del alelo HLA-A3 del complejo mayor de histocompatibilidad en el cromosoma 6.
Uno de los hitos más importantes en la investigación de la hemocromatosis llegó en 1996 cuando John N. Feder y colaboradores identificaron el gen HFE y las dos principales mutaciones asociadas a él en la enfermedad (C282Y y H63D).[5]
En el año 2000 se descubrió que un grupo de pacientes no presentaba las mutaciones descritas en 1996 ni ninguna otra asociada al gen HFE. El resto de genes implicados se identificaron en los 4 años siguientes. A partir del año 2004, debido a las nuevas investigaciones, se restableció la clasificación de las hemocromatosis.
Epidemiología
La hemocromatosis hereditaria es la enfermedad genética más frecuente en Occidente, donde afecta a 1 de cada 200 personas.[6] Todos los estudios han demostrado su mayor prevalencia en individuos de raza blanca, de origen caucásico y céltico. La migración de los pueblos celtas explicaría, quizá, la diseminación de la enfermedad por todo el mundo.
Además de las diferencias étnicas, el sexo también parece guardar relación con la enfermedad (sin ser una patología de herencia ligada al sexo). Los hombres la padecen con mayor frecuencia que las mujeres, en una proporción 3:1.[7] Factores fisiológicos femeninos, como son la menstruación o el embarazo, impiden grandes acúmulos de hierro en el organismo, siendo ésta una de las causas de dicha discordancia.
Etiología y clasificación
Existen dos formas en que se puede presentar la enfermedad:
- Hemocromatosis hereditaria o primaria: se debe a una alteración genética.
- Hemocromatosis adquirida o secundaria: distintas etiologías (patologías hematológicas, especialmente) conducen a su génesis.
Hemocromatosis hereditaria o primaria
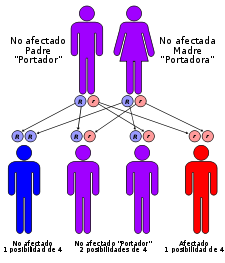 Ejemplo de herencia autosómica recesiva.
Ejemplo de herencia autosómica recesiva.Las causas de la enfermedad hereditaria son mutaciones en el gen HFE, localizado en el cromosoma 6.[8] El HFE codifica para una proteína (perteneciente a la familia de moléculas del sistema mayor de histocompatibilidad HLA-A) que participa en la regulación de la absorción del hierro y se expresa, en niveles altos, en órganos como el hígado y el intestino delgado.[7] [9] Se trata de mutaciones puntuales, donde dos pares de bases complementarias del ADN se intercambian:
- Mutación C282Y: la sustitución de guanina por adenina,[7] da lugar al cambio del aminoácido cisteína por tirosina, ocasionando una alteración en la proteína codificada por el gen HFE. Del 85 al 100% de los pacientes padecen esta mutación.[10]
- Mutación H63D: la citosina es sustituida por una guanina,[7] dando lugar al intercambio de histidina por ácido aspártico (con su consecuente repercusión en la proteína final). Es mucho menos frecuente.
La herencia de la enfermedad es autosómica recesiva,[8] lo que significa que para padecer este tipo de hemocromatosis es necesario que se herede el gen mutado de ambos progenitores, es decir, el paciente será homocigoto para la mutación, presentando el genotipo: C282Y/C282Y. Si sólo presenta una copia, se dice que ese sujeto es portador de la enfermedad (la puede transmitir a la descendencia pero él no la sufre). Un pequeño porcentaje de individuos homocigotos para la mutación no padece la enfermedad, dado que presentan menor penetrancia;[10] este es el caso de las mujeres, debido a las pérdidas de hierro mensuales que sufren por la menstruación. Pero existen también otros factores que pueden modificar la expresión de la enfermedad: el aporte de hierro en la dieta, el consumo de alcohol, ciertas infecciones víricas y anemias crónicas.[11]
Hay que tener en cuenta que esta homocigosis, como condicionante de la enfermedad, no se cumple con la mutación H63D: el genotipo H63D/H63D no causa hemocromatosis; es necesario que coexistan ambas (C282Y/H63D) para que se desarrolle el problema. A esta variante se la llama heterocigoto compuesto.[12]
El paciente con mutación experimenta un aumento gradual de la saturación de la transferrina y modificaciones en los niveles de la ferritina plasmática.
Las mutaciones en el gen HFE protagonizan la mayor parte de las hemocromatosis, pero existen otras formas de enfermedad en las que están implicados otros genes. En la siguiente tabla,[13] se recogen los cuatro tipos de hemocromatosis primaria:
Tipo OMIM Gen mutado Descripción Hemocromatosis tipo 1 o hemocromatosis hereditaria (HH) 235200[14] HFE: gen que codifica para la proteína que lleva su nombre, reguladora de la absorción de hierro Aumentan los niveles de hierro libre y su posterior absorción intestinal Hemocromatosis tipo 2A o hemocromatosis juvenil[15] 602390[16] HJV: codifica para la hemojuvelina, proteína moduladora de la hepcidina Depósito de hierro en los tejidos antes de los 30 años en ambos sexos Hemocromatosis tipo 2B o hemocromatosis juvenil[15] 606464[17] HAMP: gen que codifica para la proteína hepcidina El déficit de HAMP provoca sobrecarga de hierro Hemocromatosis tipo 3 604720[18] TfR2: codifica para el receptor 2 de la transferrina Clínica indistinguible de la hemocromatosis tipo 1 Hemocromatosis tipo 4 604653[19] SLC40A1: gen que codifica para la ferroportina Herencia autosómica dominante. Clínica similar a hemocromatosis tipo 1 Hemocromatosis tipo 3
El receptor de la transferrina 2 (TfR2) es una glicoproteína transmembrana cuya porción extracelular se une a la transferrina que transporta el hierro.[20] Esta es la principal vía de entrada de hierro en las células y se localiza, sobre todo, en el hígado y en las células de las criptas del duodeno. La clínica de estos pacientes es muy similar a la manifestada en la hemocromatosis tipo 1, pero suele aparecer en edades más jóvenes.[20]
Hemocromatosis adquirida o secundaria
La causa de la enfermedad secundaria es el aumento de los depósitos de hierro del organismo a consecuencia de diferentes hechos:[21]
- Múltiples transfusiones sanguíneas: las anemias crónicas (ferropénicas o talasemias), iatrogénicas y una eritropoyesis ineficaz requieren, en algunas ocasiones, de aporte sanguíneo y, con ello, grandes cantidades de hierro para su restitución.
- Anemia hemolítica: la destrucción de los eritrocitos conlleva la liberación del hierro de su interior. Al encontrarse éste fuera del glóbulo rojo, tiene mayor tendencia a acumularse en los tejidos.
- Enfermedades hepáticas: como son la hepatitis C y la hepatopatía alcohólica.
- Alcoholismo crónico: afecta en gran medida al hígado. Bebidas como el vino, con un alto contenido de hierro,[22] favorecen el aumento de la transferrina.
- Déficit congénito de transferrina: la ausencia (o déficit) de la proteína transportadora de hierro, favorece la acumulación del metal en los órganos y cursa con una anemia hipocrómica microcítica que mejora con la transfusión de transferrina.[23]
- Aceruloplasminemia: una mutación en el gen de la ceruloplasmina hace que el hierro se deposite en el hígado, en el sistema nervioso central y en los ojos, originándose síntomas neurológicos extrapiramidales, cerebelosos y oculares.[23]
- Ingesta excesiva de hierro: el consumo elevado de alimentos con mucho hierro saturan el proceso de absorción intestinal.[24]
- Porfiria cutánea tarda: se ha demostrado que pacientes porfirínicos son portadores de la mutación de la hemocromatosis.[25] Suelen tener un acúmulo de hierro en el hígado.
- Complicación en la derivación porto-cava: tratamiento quirúrgico en la hipertensión portal en que se anastomosan las venas porta y cava inferior.[21]
- Sobrecarga de hierro africana (también llamada siderosis de los bantúes):[26] debe el nombre a su distribución geográfica en África subsahariana. Hay dos teorías que intentan explicar su etiología; por una parte, el exceso de hierro ingerido mediante la toma de una bebida de maíz que se fermenta en recipientes hechos de este metal y, por otra parte, un gen no relacionado con el sistema HLA. En esta enfermedad los depósitos de hierro se dan en el sistema fagocítico mononuclear y en las células parenquimatosas.[23]
- Hemocromatosis neonatal: su característica principal es una insuficiencia hepática grave que comienza cuando el feto está dentro del útero, donde el acúmulo de hierro es en las células del parénquima y escasamente en el sistema fagocítico mononuclear.[23] La causa puede ser una cierta exposición ambiental intraútero o una herencia mitoconcrial, dado que hay riesgo de que se repita en embarazos posteriores.[27]
Radicales libres
Diversos estudios han concluido que altas cantidades de hierro pueden contribuir a aumentar los radicales libres de oxígeno en el organismo.[28] El motivo es la rápida oxidación que sufre este metal y su reacción con moléculas orgánicas para formar especies reactivas de oxígeno que aumentan el estrés oxidativo, el cual resulta nocivo para el cuerpo. El daño es consecuencia del acúmulo de estos radicales en diversos órganos como son el hígado, el páncreas, el corazón, las articulaciones y la piel, e incluso, las neuronas, llevando a la enfermedad de Alzheimer, por ejemplo. También tiene que ver con la disfunción endotelial.[28]
En relación con los radicales libres algunos estudios hablan acerca de la relación de la vitamina C y los daños oxidativos que puede ocasionar en el individuo. El ácido ascórbico favorece la absorción intestinal de hierro, lo que es beneficioso para la prevención de la anemia ferropénica; sin embargo, no es bien sabido si también aumenta dicha absorción en personas con sobrecarga férrica.[29] Los estudios avalan que grandes cantidades de vitamina C no producen desequilibrios en la absorción de hierro ni en personas sanas ni en heterocigotos para la mutación del gen HFE,[29] pero no existen evidencias contrastadas para los homocigóticos. Se ha podido demostrar que in vitro, la vitamina C se comporta como un pre-oxidante en presencia de metales de transición, como es el caso del hierro, pero in vivo sus conocidas funciones son como uno de los principales antioxidantes con los que cuenta el organismo humano.[29]
Patogenia
 Distribución del hierro en el organismo.
Distribución del hierro en el organismo.La cantidad de hierro en el organismo humano es de 4 a 5 gramos, distribuido éste en la hemoglobina (65% del hierro total), en el sistema retículo endotelial e hígado (15 - 30%), en la mioglobina (4%) y en diversos sistemas enzimáticos.[30] Cada día se pierde alrededor de 1 mg de hierro (cantidad superior en las mujeres por las pérdidas menstruales) y los requerimientos dietéticos diarios deben ser suficientes para compensar el excretado. Este equilibrio está roto en la hemocromatosis, donde la absorción intestinal de hierro se ve muy aumentada. Hay dos mecanismos que explican cómo tiene lugar este desorden homeostático: mediante el papel de las criptas intestinales y de la hepcidina.
Criptas intestinales
El hierro es absorbido en los enterocitos de las criptas del intestino delgado gracias a la actuación coordinada de una proteína divalente de metales (DMT-1) y de la ferroportina 1, expresadas ambas en estas células intestinales.[31] Por otra parte, la transferrina (Tf) se asocia a las membranas celulares, especialmente de los hepatocitos, interaccionando por medio del receptor de la transferrina (TfR), para depositar el hierro que transporta en sangre. Dicha asociación está, en parte, regulada por la HFE. Estudios histoquímicos han localizado esta proteína en situación intracelular y perinuclear de los enterocitos de las criptas, pero con distinta situación en los hepatocitos.[32] Por eso se distinguen las diferentes funciones que ejecuta la proteína HFE en ambas células, para poder entender cuál es su mecanismo patogénico en la hemocromatosis:
- La HFE normal capacita a los enterocitos para detectar la cantidad de hierro sanguíneo, permitiendo que entre en ellos. Cuando la proteína HFE sufre una mutación, no se detecta la cantidad de hierro corporal, éste no entra en el enterocito y, por consiguiente, se absorbe una mayor cuantía.[33]
- En condiciones normales, la HFE se asocia en la membrana de los hepatocitos con el TfR disminuyendo la afinidad de éste por la Tf; de esta manera, evita que el hierro se deposite en el hígado. Sin embargo, la HFE mutante pierde esta capacidad inhibitoria sobre la unión TfR-Tf y el hierro es acumulado en el órgano. Existen diferencias entre las mutaciones que puede albergar la proteína: mientras que la variación C282Y impide su llegada al hepatocito, la H63D permite que llegue, pero interfiere en su correcta función.[32] El resultado en ambos casos es el mismo: una hemocromatosis por sobrecarga férrica en el hígado.
Hepcidina
La hepcidina es un péptido hepático que trabaja a nivel de los enterocitos, del hígado y de los macrófagos, regulando la liberación de hierro a la sangre. En condiciones fisiológicas, un aumento de hierro sérico estimula la secreción del péptido, que inhibe el transporte del metal desde el enterocito (mediante su interacción con la ferroportina), hígado o sistema retículo endotelial, a la sangre.[34] Este sistema no funciona en la hemocromatosis: estudios con ratones knockout, homocigotos para la mutación del gen HFE,[34] presentan una deficiencia de hepcidina, lo que explica esa sobrecarga de hierro incontrolada en estos pacientes. También se ha descubierto una mutación en el gen que codifica para la ferroportina,[35] confiriéndole resistencia frente a la hepcidina. El resultado vuelve a ser el mismo: un incremento de hierro en el organismo.
Cuadro clínico
Existen dos puntos comunes a cualquier tipo de hemocromatosis: la hemosiderosis generalizada y la afección orgánica (suele ser cirrosis o fibrosis hepática).[27] Además del hígado, que es el órgano más afectado en los pacientes hemocromatosos, existen otros tejidos que sufren la sobrecarga férrica: páncreas, corazón, órganos endocrinos (hipotálamo, hipófisis, tiroides), articulaciones y piel.
La alteración de cada uno de estos órganos, que será variable en cada paciente, es lo que determina la clínica; si bien, aunque el defecto genético (en el caso de la HH) existe ya desde el nacimiento, los signos y síntomas se revelan en la edad adulta cuando la demasía de hierro es más agresiva (hasta entonces la enfermedad era asintomática). De esta forma, la evolución de la hemocromatosis se divide en 3 estadios:[7]
- Estadio I: no hay sobrecarga de hierro.
- Estadio II: hay sobrecarga, pero no morbilidad. En esta fase son protagonistas síntomas muy inespecíficos, como el cansancio o el letargo.
- Estadio III: hay sobrecarga de hierro y morbilidad clínica. Se manifiesta la sintomatología clínica correspondiente a la afección orgánica y, para ello, la cantidad mínima necesaria acumulada en éstos tiene que ser superior a 20 gramos.[36]
Afección hepática
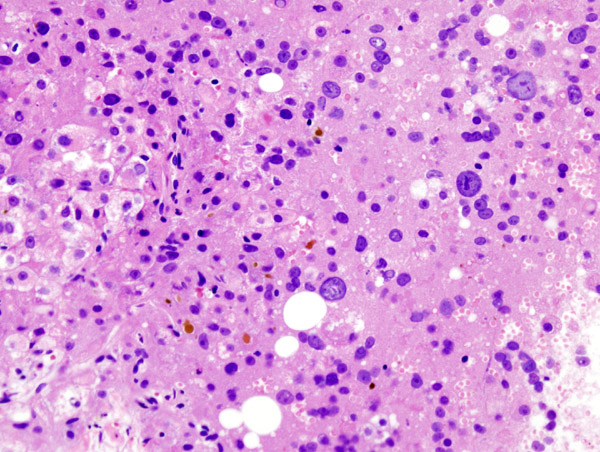
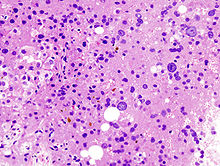 Imagen histológica de un hepatocarcinoma en un paciente con cirrosis hepática.
Imagen histológica de un hepatocarcinoma en un paciente con cirrosis hepática.Como ya se ha dicho anteriormente, el hígado es el órgano que resulta más afectado ante el exceso de hierro. Histopatológicamente, el metal se deposita en los hepatocitos, células de Kupffer (macrófagos hepáticos) y en las células del epitelio biliar, principalmente.[37] A consecuencia de ello, se pueden originar una fibrosis hepática que, si evoluciona, desencadena en una cirrosis (se da en el 60% de los pacientes),[4] y un hepatocarcinoma, porque aumenta la susceptibilidad del hígado a desarrollar tumores malignos (en un 5% de los casos).[4] Por otra parte, un signo claro en las hemocromatosis es la hepatomegalia, con predominio del lóbulo izquierdo, y su firme consistencia a la palpación;[23] otros menos comunes son la esplenomegalia, la ascitis, el edema y la ictericia.
Afección pancreática
Aproximadamente, un 50 ó 60% de las personas que sufren hemocromatosis presenta diabetes.[11] Esta manifestación puede tener doble etiología: por un lado, la sobrecarga de hierro existente en las células beta del páncreas endocrino (encargadas de secretar la insulina); y, por otro lado, la resistencia a la insulina por lesión hepatocelular.[38] Este último caso hace que, en muchas ocasiones, los pacientes con hemocromatosis sean diagnosticados como enfermos de diabetes mellitus tipo 2.
Daño cardíaco
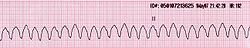 Taquicardia ventricular en un paciente con hemocromatosis (Derivación II del ECG).
Taquicardia ventricular en un paciente con hemocromatosis (Derivación II del ECG).El acúmulo excesivo de hierro en las fibras musculares cardíacas puede dar lugar a dos trastornos diferentes que aparecen en un tercio de los pacientes:[39] por un lado, una insuficiencia cardíaca congestiva (del 15 al 35% de los casos),[4] con disfunción sistólica y diastólica, manifestada mediante cansancio, astenia y edema en zonas declives (pies); y por otro, unas alteraciones en el ritmo cardíaco (arritmias) (del 20 al 30% de frecuencia),[4] como son las bradicardias, las taquicardias auriculares y/o ventriculares y las extrasístoles.
En estos pacientes existe un riesgo de muerte por miocardiopatía 300 veces mayor que en el resto de la población.[11] Esto se debe a que la ferritinemia alta (ferritina plasmática) está asociada a un incremento del riesgo de sufrir una enfermedad coronaria.[11]
Endocrinopatía
La afección del hipotálamo y de la hipófisis da lugar a diferentes manifestaciones endocrinológicas (presentes en el 15-35% de pacientes):[4]
- Hipogonadismo secundario: se expresa, sobre todo, en los hombres acompañado de atrofia testicular, pérdida de la libido e impotencia sexual. Su causa es el déficit en la secreción de gonadotropinas por parte del eje hipotálamo-hipofisario.[40] Es muy poco frecuente que en la mujer cause amenorrea.
- Hipotiroidismo secundario: el acúmulo de hierro interfiere en la secreción hipofisaria de TSH,[41] lo que dificulta la producción de hormonas tiroideas por la glándula tiroides.
- Hipoparatiroidismo secundario: de igual modo que en los dos casos anteriores, el exceso de hierro repercute negativamente en la secreción de PTH y, por tanto, las glándulas paratiroides son hipofuncionales.
Artropatía
La afección de las articulaciones es una manifestación común que ataca del 30 al 40% de los pacientes.[4] Las alteraciones tienen lugar a nivel de tres estructuras: la membrana sinovial, el cartílago y el hueso.
- Membrana sinovial: dos son las agresiones que la dañan. Por una parte, los acúmulos desproporcionados de pigmento hemosiderínico en los sinoviocitos (células que componen la membrana sinovial); y por otra parte, los depósitos de cristales de pirofosfato cálcico que ocasionan ataques recurrentes de artritis aguda o pseudogota.
- Cartílago articular: aquí también los depósitos de pirofostafo cálcico deterioran el cartílago.
- Hueso: sufre un engrosamiento de sus trabéculas, unido a la infiltración celular en la zona (fruto de la presencia de pirofosfato cálcico) y a la proliferación vascular del tejido conectivo próximo al cartílago erosionado.
Esta osteoartritis degenerativa es progresiva y crónica, generalmente simétrica. Principalmente, las articulaciones que sufren daños son la segunda y tercera metacarpofalángicas; con menos frecuencia, encontramos también artropatía en las articulaciones de la muñeca, rodilla, hombro y cadera.[11]
Alteraciones de la piel
Aproximadamente el 70% de los pacientes presenta una tonalidad más oscura de la piel (hiperpigmentación).[4] A menudo se aplica el término diabetes bronceada a la hemocromatosis cuando presenta la triada de cirrosis, pigmentación y diabetes; fue Charles Emile Troisier quien le dio esta denominación, hasta que von Recklinghausen, en 1889, la sustituyó por hemocromatosis.[42] (Véase Historia)
También se pueden observar las uñas aplanadas o cóncavas y una disminución del vello corporal.
Otras manifestaciones
Los individuos con hemocromatosis también pueden presentar otros síntomas menos comunes; por ejemplo, la afección de los ganglios de la base puede ocasionar movimientos involuntarios conocidos como discinesias. Además, existe una mayor susceptibilidad a ciertas infecciones, causadas por microorganismos que se desarrollan mejor en presencia de hierro, como Yersinia enterocolitica, Listeria monocytogenes y Vibrio vulnificus.[12]
Diagnóstico
En muchas ocasiones la hemocromatosis se diagnostica mediante exámenes sanguíneos de rutina, dado que los síntomas más específicos suelen aparecer tardíamente. Para llegar a confirmar que una persona padece la enfermedad, se debe seguir un orden en la cadena diagnóstica: primero se estudia la clínica del paciente, seguida de una bioquímica que confirme la sospecha; a continuación, se realiza un test genético si la bioquímica muestra alteraciones y, finalmente, se hace una biopsia hepática que detalla el daño sufrido en el hígado.
Sospecha clínica de hemocromatosis
Ciertas manifestaciones patológicas orientan al clínico hacia una posible alteración metabólica: astenia, diabetes, hepatopatía crónica, artralgias, miocardiopatías o impotencia, entre otras.[43] Una buena historia clínica, con una completa anamnesis y exploración exhaustiva, ayuda al médico en el diagnóstico. Sin embargo, dado que la mayor parte de los pacientes son asintomáticos durante mucho tiempo, la sospecha de hemocromatosis viene iniciada por el resultado de las pruebas bioquímicas.
Bioquímica
Hay tres parámetros que son de gran utilidad en el diagnóstico:
- Saturación de transferrina (entre 30 y 45%, en condiciones normales):[7] se considera anormal un valor superior al 45% o al 55%,[44] en mujeres u hombres respectivamente. Este valor no es proporcional a la sobrecarga férrica.
- Ferritina sérica (entre 20-300 ng/ml en el hombre y entre 20-200 ng/ml en la mujer, en situación fisiológica): cifras por encima de los 350 ng/ml se consideran altas. A diferencia de la determinación anterior, la ferritina es proporcional a la cuantía de depósito de hierro en el organismo, aunque tarda más en alterarse.
- Hierro sérico (los valores de referencia son de 50 a 150 mcg/dl en el hombre y de 35 a 145 mcg/dl en la mujer): cuando las cifras superan los 150 mcg/dl hacen sospechar de una posible alteración metabólica; sin embargo, dado que el nivel de hierro en sangre puede verse afectado por multitud de variables, como son la ingesta o situaciones inflamatorias, su uso como marcador de hemocromatosis es muy limitado.
Análisis genético
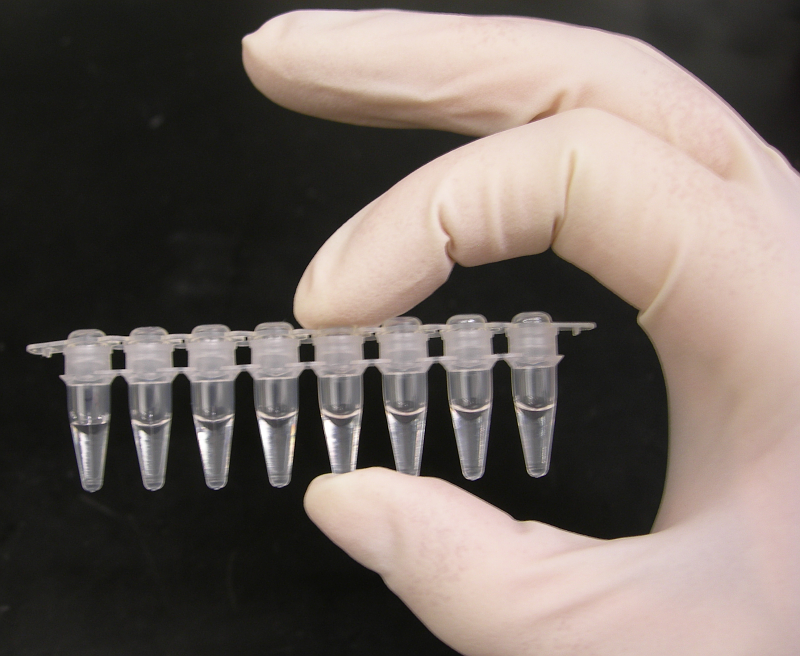
 Tubos de PCR con muestra de ADN.
Tubos de PCR con muestra de ADN.Tras encontrarse alteraciones en las pruebas bioquímicas (especial importancia tiene la saturación de transferrina superior al 45%) se procede a realizar al paciente una prueba de ADN para encontrar las mutaciones C282Y y/o H63D y, finalmente, confirmar la enfermedad. Se trata de un análisis genético basado en la amplificación de un fragmento del ADN del paciente mediante la reacción en cadena de la polimerasa (PCR) y posterior identificación de la mutación con la ayuda de enzimas de restricción y una electroforesis. Para diagnosticar la hemocromatosis es necesario que el test detecte la homocigosis C282Y/C282Y o la heterocigosis compuesta C282Y/H63D. Un resultado negativo para ambas mutaciones no excluye de enfermedad, sino que obliga al paciente a someterse a un seguimiento y a un completo estudio de su historia clínica; además deben ser descartadas otras causas de enfermedad antes de poder decir que no padece hemocromatosis. Este análisis presenta una fiabilidad de más del 99%.[10]
Biopsia hepática
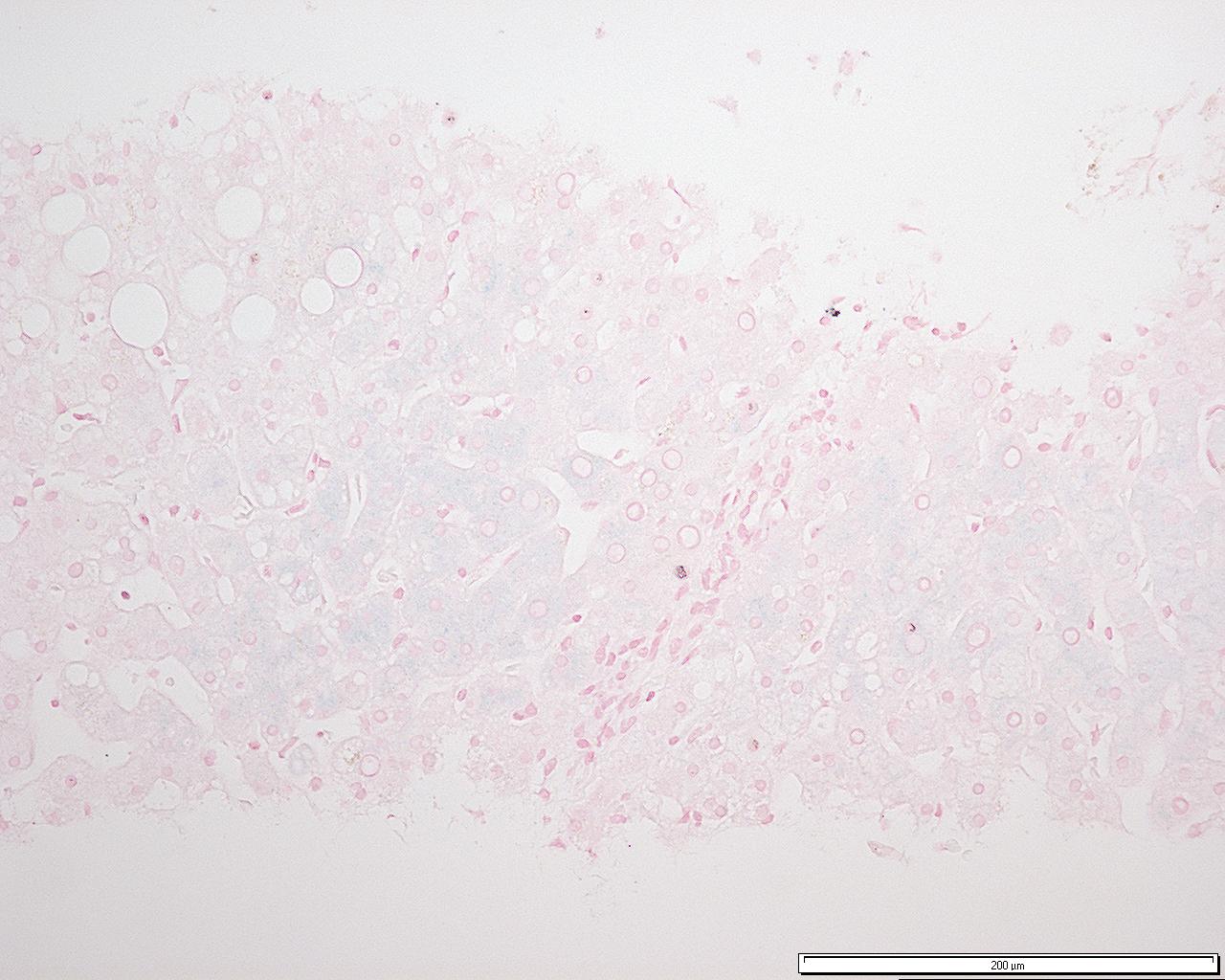
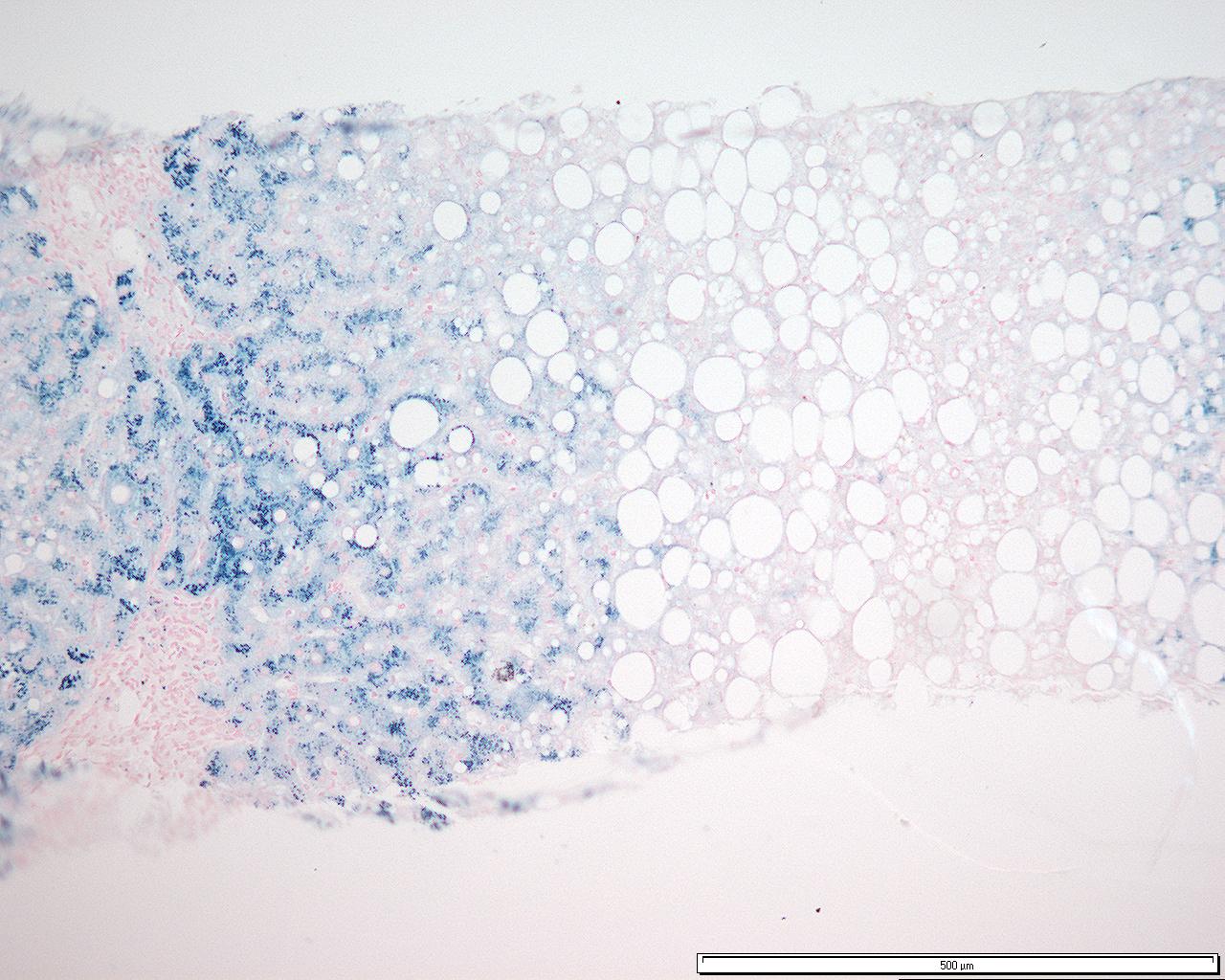
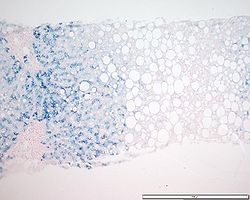 Acumulación de hierro de grado 3 en hepatocitos que muestra un patrón de distribución acinar consistente con una hemocromatosis genética homocigótica (preparado con tinción azul de Prusia de Perl).
Acumulación de hierro de grado 3 en hepatocitos que muestra un patrón de distribución acinar consistente con una hemocromatosis genética homocigótica (preparado con tinción azul de Prusia de Perl).Es el método diagnóstico más invasivo y el que determina la cuantía de la sobrecarga, así como el grado de lesión histológica (sobre todo el intervalo entre fibrosis y cirrosis), el pronóstico de la enfermedad y descarta otras causas como hepatitis viral o daño por sobrecarga etílica.
Indicaciones de la biopsia
Existen dos supuestos principales en los que se recurre a la biopsia hepática: uno son los pacientes con ferritina superior a 1000 ng/ml,[45] aumento de las transaminasas o hepatomegalia; y otro, los pacientes con test genético negativo para las mutaciones del gen HFE, pero que mantienen niveles de saturación de la transferrina por encima del 45% y ferritina superior a 300 ng/ml.[45] Los individuos con resultado positivo para la homocigosis o heterocigosis antes descritas en el test genético, no se someten a la biopsia porque su diagnóstico de hemocromatosis ya está establecido (a no ser que haya sospecha de daño hepático y se quiera verificar el mismo).
Imagen histológica
Cuando hay sobrecarga de hierro el resultado de la biopsia muestra grandes cantidades en el órgano, pero existen unas diferencias de localización según si dicha sobrecarga es primaria o secundaria: en la hemocromatosis primaria las grandes cantidades de hierro se acumulan en los hepatocitos, mientras que en la hemocromatosis secundaria el depósito se origina primero en las células de Kupffer y, posteriormente también, en las células hepáticas.[45]
La fibrosis en el hígado aparece con unos niveles más elevados de hierro y tiene una distribución inicial periportal para, más adelante, organizarse en puentes portoportales. Finalmente aparece la cirrosis mixta en forma de macro y micronódulos.[46]
Tratamiento
El tratamiento de la hemocromatosis busca la deplección de los depósitos de hierro en exceso. La terapia difiere según la causa de la enfermedad; así en las hemocromatosis primarias el tratamiento de elección es la flebotomía mientras que, en las secundarias, se opta por los quelantes de hierro. Sólo en aquellos casos en que surgen complicaciones hepáticas se plantea la posibilidad de un trasplante. También es importante el tratamiento dietético que debe seguir el paciente y la toma de ciertos fármacos que tratan el resto de síntomas derivados de la enfermedad.[47]
Flebotomías
Con ellas se extrae sangre al sujeto regularmente, para disminuir esa sobrecarga de hierro. El tratamiento inicial consiste en flebotomías semanales de 500 ml de sangre completa (250 mg hierro/ 500 ml de sangre),[48] durante 2 o 3 años. En los primeros meses, hay un descenso de la ferritina sérica, tardando más en menguar los valores de la saturación de transferrina y la sideremia (cantidad total de hierro en sangre), que necesitan un descenso de ferritina sérica a 50 ng/ml para que se aprecie su disminución; en ese momento también la hemoglobina baja, por ello es uno de los principales valores control que se emplea para saber la evolución del tratamiento.
Cuando la saturación de transferrina y la ferritina se sitúan por debajo del 50% y de 50 ng/ml respectivamente, se sigue con la siguiente etapa de tratamiento: flebotomías de mantenimiento; en éstas se aumentan los intervalos de aplicación de una semana a 3 o 4 meses.[48] En muchas ocasiones esta etapa suele ser crónica.
Las flebotomías, al bajar el depósito férrico, regresan la hepatomegalia, la debilidad, la astenia y aseguran un mejor control de la diabetes. Esto no es así en pacientes que han desarrollado ya cirrosis; en ellos mejora la calidad de vida.
Quelantes de hierro
Son el tratamiento de elección en las sobrecargas de hierro secundarias. Los quelantes son moléculas que se unen al hierro formando compuestos solubles y, de esta manera, evitan que el metal ejerza su efecto tóxico en el organismo. En la actualidad existen tres quelantes que pueden emplearse en el tratamiento de esta enfermedad:
- Deferoxamina: es el quelante más antiguo que se utiliza e intenta frenar la evolución de la enfermedad y la muerte prematura por dicha causa. Se administra, especialmente, por vía subcutánea, ya que presenta escasa biodisponibilidad oral. La forma de administración y la frecuencia de la misma (cada 8 - 12 horas de 3 a 7 días a la semana),[49] pueden provocar que el paciente acabe abandonando el tratamiento a los pocos meses de su inicio.
- Deferiprona: está aprobado su uso en Europa, como alternativa al fracaso con deferoxamina o como tratamiento combinado. Su ventaja frente al primero es su fácil administración, pero su efecto es menos eficaz y se ha relacionado con casos de leucopenia en algunos pacientes.[49]
- Deferasirox: el Ministerio de Sanidad ha aprobado su uso en España.[49] Se administra por vía oral una vez al día y, debido a sus efectos secundarios (náuseas, vómitos, diarrea, dolor abdominal, exantema, trastornos oculares y auditivos, entre otros), debe hacerse bajo un riguroso control médico.
Otros fármacos
Del exceso de hierro se derivan muchos síntomas que no todos desaparecen con las flebotomías o los quelantes; son necesarios fármacos específicos que controlen dichos problemas: la testosterona es útil para luchar contra la impotencia, el hipogonadismo y la pérdida de vello;[48] los antiinflamatorios no esteroideos (AINEs) se dan para la artropatía, que es uno de los principales problemas que soportan estos pacientes y se consigue disminuir el dolor en las uniones de los huesos.[48] Una técnica más agresiva es la artroplastia, cirugía en que se reemplazan las articulaciones;[50] por último, también son importantes los hipoglucemiantes para tratar la diabetes ocasionada y diversos fármacos que corrigen las miocardiopatías.[50]
Tratamiento dietético
Además de la correspondiente terapia, el sujeto debe llevar a cabo un correcto régimen dietético que lo ayude a mantener bajos los niveles de ferritina en sangre:[50] es importante que elimine el consumo de alcohol porque el etanol puede agravar el daño hepático; hay que limitar el consumo de carne roja, por su alto contenido en hierro; no se deben consumir pescados ni mariscos crudos, pues pueden contener ciertos microbios frente a los que estos pacientes son más susceptibles; hay que evitar la toma de suplementos vitamínicos que contengan hierro y, por último, es conveniente limitar el aporte de vitamina C, ya que aumenta la cantidad de hierro que se ingiere con los alimentos.
Trasplante hepático
Cuando existe una complicación hepática grave, como es la insuficiencia hepática, se inicia el protocolo de trasplantes. El riesgo de muerte en estos pacientes es mayor que en un cirrótico sin hemocromatosis previa pues intervienen diversas variables añadidas, como una mayor frecuencia de infección o la miocardiopatía añadida por el exceso de hierro.
Pronóstico
El pronóstico será más favorable cuanto antes se diagnostique la enfermedad, se inicie el tratamiento y menos daño hístico exista.
En general, estos sujetos tienen una esperanza de vida menor que el resto de la población sana de la misma edad. Las principales causas de mortalidad son (por orden decreciente de frecuencia): la insuficiencia cardiaca congestiva, la insuficiencia hepática, la hipertensión portal y el carcinoma hepatocelular.[51]
No obstante, la calidad de vida del paciente también está disminuida. La cirrosis, la diabetes y, sobre todo, la artropatía derivada influyen negativamente en la forma de vivir del individuo.
Referencias
- ↑ «What Is Hemochromatosis?» (en inglés).
- ↑ «Diferenciación de hemosiderosis y hemocromatosis». Consultado el 17-09-2008.
- ↑ Galleano, Mónica; Puntarulo, Susana. «El hierro y su relación con los radicales libres». Universidad de Buenos Aires. Consultado el 17-09-2008.
- ↑ a b c d e f g h «Síntomas y signos más frecuentes. Introducción histórica». Consultado el 17-09-2008.
- ↑ «Genética molecular de la hemocromatosis hereditaria». Consultado el 17-09-2008.
- ↑ Dra. Masciovecchio. «Aspectos básicos de la hemocromatosis». Consultado el 17-09-2008. «1 de cada 8 caucásicos porta el gen.».
- ↑ a b c d e f «El gen HFE y sus mutaciones». Consultado el 17-09-2008.
- ↑ a b Ferri, Fred F (2006). «Enfermedades y trastornos: Hemocromatosis». Ferri consultor clínico 2006-2007: Claves diagnósticas y tratamiento. Elsevier España. ISBN 8481749141. «Véase página 390.»
- ↑ «Mutaciones del gen HFE». Consultado el 17-09-2008. «El 90% de las personas con hemocromatosis presenta la mutación C282Y.».
- ↑ a b c «Análisis genético para la hemocromatosis hereditaria». Consultado el 17-09-2008.
- ↑ a b c d e «Factores que interfieren en la expresión de la mutación». Consultado el 17-09-2008.
- ↑ a b «Heterocigosis compuesta y manifestaciones clínicas». Consultado el 17-09-2008.
- ↑ «Tipos de hemocromatosis». Consultado el 17-09-2008. «Los datos de la tabla se han obtenido de la presente página.».
- ↑ «Hemocromatosis hereditaria (HFE)» (en inglés). OMIM. Consultado el 11-01-2009.
- ↑ a b «Variantes del tipo 2». Consultado el 17-09-2008. «La hemocromatosis 2A y 2B son subtipos de la hemocromatosis tipo 2.».
- ↑ «Hemocromatosis juvenil (HJV)» (en inglés). OMIM. Consultado el 11-01-2009.
- ↑ «Hepcidin Antimicrobial Peptide (HAMP)» (en inglés). OMIM. Consultado el 11-01-2009.
- ↑ «Transferrin Receptor 2 (TfR2)» (en inglés). OMIM. Consultado el 11-01-2009.
- ↑ «Solute Carrier Family 40 (SLC40A1)» (en inglés). OMIM. Consultado el 11-01-2009.
- ↑ a b «Características específicas de los distintos tipos de hemocromatosis». Consultado el 21-2-09.
- ↑ a b «Etiología de la hemocromatosis secundaria». Consultado el 17-09-2008.
- ↑ Morgan, Sarah L. (1999). «Enfermedades de la sociedad moderna». Nutrición Clínica. Elsevier España. ISBN 8481744662.
- ↑ a b c d e Rodes, J (2001). «Trastornos metabólicos hereditarios». Tratado de hepatología clínica. Elsevier España. ISBN 8445809660. «Véanse páginas 1558 y 1564.»
- ↑ «Lista de alimentos ricos en hierro». Consultado el 17-09-2008.
- ↑ Wolff F, Carlos; Armas M, Rodolfo; Frank, Jorge et al (2006). «Mutaciones del gen de la Hemocromatosis en donantes de sangre voluntarios y en pacientes con Porfiria cutánea tarda en Chile». Medicina (B.Aires) 66 (5). 0025-7680 , 421-426. http://www.scielo.org.ar/scielo.php?script=sci_arttext&pid=S0025-76802006000500006&lng=es&nrm=iso.
- ↑ «La sobrecarga de hierro africana» (en inglés). Consultado el 07-02-2009.
- ↑ a b Kumar, Vinay; Cotran, Ramzi S.; Robbins Stanley L. (2003). «Lesión, adaptación y muerte celular». Patología humana: Robbins (Séptima edición). Elsevier España. ISBN 8481746665. «Véase página 20 y 616.»
- ↑ a b «La hemocromatosis y otras enfermedades asociadas» (en inglés). Consultado el 13-12-2008.
- ↑ a b c Gerstcr, H (1999). «High-dose Vitamin C: A Risk for Persons with High Iron Stores?». International Journal for Vitamin and Nutrition Research 69. 10.1024/0300-9831.69.2.67, p.67-82. http://verlag.hanshuber.com/Zeitschriften/IJVNR/99/vn9902.html.
- ↑ Guyton, Arthur C.; Hall, John E. (2006). «Eritrocitos, anemia y policitemia». Tratado de Fisiología médica (Décimoprimera edición). Elsevier. ISBN 8481749265. «Homeostasis del hierro»
- ↑ «Las criptas intestinales». Consultado el 17-09-2008. «Papel de la DMT-1 y la ferroportina en la absorción del hierro.».
- ↑ a b Oliva, Rafael; Ballesta, Francisca; Oriola, Josep; Clària, Joan. «Enfermedades autosómicas recesivas». Genética médica. Edicions Universitat Barcelona.
- ↑ Tapias, Mónica. «Modelo de las criptas intestinales». Hospital Universitario Fundación Santa Fe de Bogotá. Consultado el 17-09-2008.
- ↑ a b «Estudio de la hepcidina en la hemocromatosis». Consultado el 17-09-2008.
- ↑ «La hepcidina como "la gran dama de hierro"». Consultado el 17-09-2008. «La hepcidina como reguladora del nivel de hierro.».
- ↑ Pardo Mindan, F.J. (1996). «Patología del metabolismo». Anatomía patológica. Harcourt. ISBN 8481741736.
- ↑ «Fisiología del hígado». Consultado el 17-09-2008. «El papel de las células de Kupffer en la protección hepática.».
- ↑ Dra. Sydney González. «Resistencia a la insulina y medicina sistémica». Consultado el 17-09-2008.
- ↑ Hospital Universitario de Salamanca (Servicio de Cardiología). «Taquicardia ventricular y hemocromatosis cardíaca.». Consultado el 17-09-2008. «Caso clínico de paciente con hemocromatosis secundaria a causa de transfusiones por anemia sideroblástica.».
- ↑ Rodríguez Sánchez, MªD. «Las causas del hipogonadismo secundario en el varón». Consultado el 17-09-2008.
- ↑ MedlinePlus. «Hipotiroidismo secundario». Consultado el 17-09-2008.
- ↑ «Diabetes coincidiendo con una enfermedad hepática». Consultado el 17-09-2008. «Denominaciones de la hemocromatosis.».
- ↑ Del Castillo Rueda, A; López-Herce Cid, J.A; De Portugal Álvarez, J. «Diagnóstico clínico de la hemocromatosis». Consultado el 17-09-2008.
- ↑ «Diagnóstico de la hemocromatosis». Consultado el 17-09-2008. «Valores bioquímicos y significado del test genético.».
- ↑ a b c «Biopsia hepática». Consultado el 17-09-2008.
- ↑ Rustgi, Anil K; Reddy, K. Rajender; Long, William B. (2005). «Trastorno del metabolismo de los metales». Los requisitos en gastroenterología. Conducto hepatobiliar y páncreas. Elsevier. ISBN 8481748226. «Véase página 777»
- ↑ MedlinePlus. «Alternativas terapéuticas». Consultado el 17-09-2008.
- ↑ a b c d «Tratamiento de la hemocromatosis». Consultado el 17-09-2008. «Terapia de la flebotomía y de las complicaciones extrahepáticas».
- ↑ a b c «Cómo se trata el exceso de hierro». Consultado el 17-09-2008. «Asociación española de hemocromatosis.».
- ↑ a b c «Cambios en la dieta y medicamentos». Consultado el 17-09-2008. «Allina. Hospitals & Clinics».
- ↑ Casseus, C.; De Campo, F.; Videla, F.; De la Vega, M.. «Causas de muerte». Consultado el 29-12-2008.
Bibliografía
- Farreras Valentí P., Von Domarus A., Cardellach F. Medicina interna. Elsevier España, 2005, 15ª ed. ISBN 84-8174-736-X.
- Kasper D.L., Braunwald E., Fauci A.S., Hauser S.L., Longo D.L., Jameson J.L. Harrison. Principios de Medicina interna. 2 Vols. McGraw-Hill, 2006, 16ª ed. ISBN 978-970-10-5165-8.
- Stevens A., Lowe J. Anatomía patológica. Elsevier España, 2001, 2ª ed. ISBN 84-8174-512-X.
Enlaces externos
Categorías:- Enfermedades hematológicas
- Enfermedades hereditarias
- Enfermedades metabólicas
- Hepatología
- Enfermedad de von Recklinghausen-Applebaum (forma hereditaria)



Wikimedia foundation. 2010.