- Telosoma
-
 Imagen de microscopía que muestra cromosomas preparados mediante una técnica conocida como FISH (Hibridación fluorescente in situ), en la que se observan los telómeros en amarillo.
Imagen de microscopía que muestra cromosomas preparados mediante una técnica conocida como FISH (Hibridación fluorescente in situ), en la que se observan los telómeros en amarillo.
El telosoma (del griego: ‘Τέλος’, fin y ‘σομα’, cuerpo), también llamado complejo protector o shelterina, es un complejo proteico, perteneciente al grupo de proteínas de unión a telómeros (TBPs), que se une al extremo final del ADN telomérico, que a su vez está situado en los extremos de los cromosomas de eucariotas.[1] Su núcleo está formado por seis subunidades, POT1, TRF1 y TRF2, que se unen directamente al ADN, y TIN2, TPP1 y RAP1, que cumplen otras funciones.[2] A su vez, este núcleo se une o interactúa más o menos temporalmente con un gran número de otras proteínas no específicas de telómeros, llamadas en ocasiones proteínas asociadas o accesorias al telosoma, como PINX1 o Apollo. Algunos autores creen que se trata de un punto central en una auténtica red de regulación a la que denominan "interactoma" de los telómeros.[3]
Varias unidades de shelterina (el complejo de seis subunidades) se pueden unir entre sí. Parece ser que esta situación estabiliza una estructura especial del ADN, denominada "bucle T" (T-loop, en inglés), en forma de lazo, a la que se ha implicado en la inhibición de la telomerasa.[2] Se han atribuido así mismo otras funciones al telosoma, entre ellas, la protección de los telómeros, probablemente inhibiendo la reparación del ADN, ya que de otro modo los extremos serían interpretados como rupturas intracatenarias, y, por tanto, desencadenaría una respuesta de reparación errónea, como el NHEJ. Así mismo parece que reprimen la forma en que las proteínas de señalización de daño en el ADN ATR y ATM arrestan el ciclo celular. Esto sugiere que un acortamiento intenso de los telómeros pararía la división celular. También estabiliza el final del cromosoma de forma directa, y regulan la longitud del telómero.
El telosoma es una estructura bastante conservada, aunque pueden variar las proteínas que la forman. Existen excepciones de organismos sin telosomas, como en algunos insectos, y muy notablemente en Drosophila melanogaster. En plantas, aunque existen datos de TBPs y existen homólogos de las proteínas en animales, se desconoce hasta el momento como están organizadas.[4] Algunos elementos del telosoma han sido implicados en varias afecciones, como el cáncer, el envejecimiento y la disqueratosis congénita.[5]
Contenido
Historia y problemas terminológicos
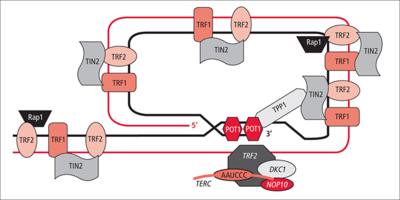 Diagrama muy esquemático de la disposición de las proteínas del telosoma de los telómeros de mamífero. En esta localización, el ADN forma un bucle grande, denominado bucle-T (T-loop). La hebra 3' del ADN termina en forma de cadena sencilla, constituyendo el "saliente" (overhang) 3' del extremo del cromosoma. La presencia de cadenas sencillas de ADN libres podría activar los sistemas de reparación, comprometiendo de esa manera la estabilidad del telómero. Para que esto no suceda, el extremo 3' invade una zona posterior de doble cadena del cromosoma, hibridando consigo misma. POT1 estabiliza esta estructura, que recibe el nombre de bucle-D (D-loop).
Diagrama muy esquemático de la disposición de las proteínas del telosoma de los telómeros de mamífero. En esta localización, el ADN forma un bucle grande, denominado bucle-T (T-loop). La hebra 3' del ADN termina en forma de cadena sencilla, constituyendo el "saliente" (overhang) 3' del extremo del cromosoma. La presencia de cadenas sencillas de ADN libres podría activar los sistemas de reparación, comprometiendo de esa manera la estabilidad del telómero. Para que esto no suceda, el extremo 3' invade una zona posterior de doble cadena del cromosoma, hibridando consigo misma. POT1 estabiliza esta estructura, que recibe el nombre de bucle-D (D-loop).El interés por el estudio de los telómeros, la región final no codificante repetitiva de los cromosomas lineales de eucariotas, radica en que se han implicado en fenómenos tan importantes como el envejecimiento o el cáncer.[6]
- Las primeras hipótesis a cerca de la naturaleza diferente del ADN del final de los cromosomas partieron de Hermann Joseph Muller (1938) y Barbara McClintock (1939).[7] El primero de ellos fue quien acuñó el término "telómero".
- El término "telosoma" se emplea aún en genética como sinónimo de cromosoma telocéntrico.[8]
- Las primeras evidencias de que la cromatina telomérica tenía un comportamiento distinto y probablemente un grupo de proteínas no nucleosomales asociadas, hablándose ya de un "complejo telomérico" vienen en 1984 con los trabajos en los protozoos Tetrahymena y Oxytricha de Gottschling y Cech.[9]
- El mismo Gottschling, trabajando en 1992 con Virginia Zakian y Jocelyn Wright descubre y caracteriza este complejo en Saccharomyces cerevisiae, y también una de las proteínas que forman parte del mismo, RAP1. Dado que el tamaño resultó ser mayor que el nucleosoma, y por semejanza con éste, acuñaron el término "telosoma" para denominarlo.[10]
- En 1999 Titia de Lange y su equipo de la Universidad Rockefeller descubrieron que el ADN telomérico posee una conformación especial a la que denomina T-Loop (Tail-loop), y que la proteína TRF2 podría ser la responsable de catalizarla.[11]
- A lo largo de la siguiente década se fueron descubriendo todos los componentes del complejo protector, y finalmente en 2004 Zou Songyang y su equipo se dieron cuenta de que, los complejos de TRF1 y TRF2, que previamente se pensaba que eran entidades separadas con distintas funciones, formaban una estructura de alto orden unida por TIN2, proponiendo restringir el término telosoma al conjunto de estas seis proteínas.[12]
- En 2005 el grupo de Titia de Lange, observando que existen muchas otras proteínas en contacto con el complejo, establece una serie de criterios para determinar cuáles de ellas pertenecen al telosoma, en especial la exclusividad de función. También propone un nuevo término, "Shelterin", cuya traducción más difundida al castellano sería "complejo protector", aunque también se emplea shelterina.[2]
- Actualmente Zhou admite la confusión terminológica refiriéndose al complejo con una mezcla de ambos términos en sus publicaciones, denominándola "Telosome/shelterin".[13]
Aislamiento
El aislamiento de la partícula telosómica se realizó por primera vez en S. cerevisiae. Para ello se aprovechó el hecho de que esta estructura protege el ADN de la proteólisis por diversas nucleasas. Una vez escindidos, se recuperan como complejos solubles de ADN-Proteína. El procedimiento hasta cierto punto similar al que se empleó para el aislamiento y estudio de los nucleosomas. A partir de la fase soluble, se verificó que la proteína RAP1 formaba parte del complejo cuando éste inmunoprecipitaba empleando anticuerpos contra RAP1. También se verificó que la zona periférica es altamente accesible a enzimas.[10]
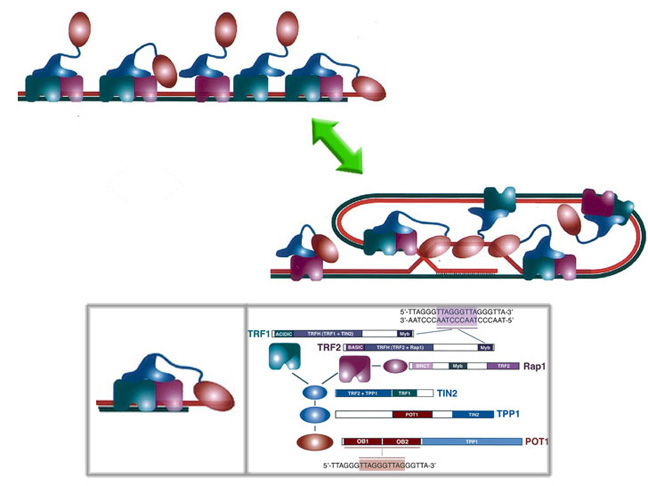
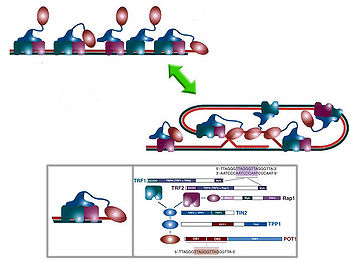 Modelo del telosoma según el grupo de Titia de Lange. Se muestra la forma en que se unen al ADN telomérico, así como los principales dominios de cada proteína.
Modelo del telosoma según el grupo de Titia de Lange. Se muestra la forma en que se unen al ADN telomérico, así como los principales dominios de cada proteína.El desarrollo de la proteómica ha variado en gran medida las técnicas de aislamiento y estudio:
- Habitualmente se ha empleado la inmunoprecipitación con anticuerpos contra la versión humana de RAP1 (hRAP1) y por supuesto, los otros componentes, en el aislamiento de telosomas humanos para experimentos a pequeña escala. A mayor escala se han utilizado geles SDS-PAGE y posteriormente se han identificado los fragmentos mediante espectrometría de masas.[12]
- Actualmente se vienen ensayando refinamientos de estas técnicas espectrométricas que además ofrecen un alto rendimiento, como la espectrometría de masas de alta resolución con cuantificación "label free", es decir, sin marcado. Un protocolo que implica todas estas técnicas ha conseguido aislar 62 proteínas de unión a telómeros, incluidas todas las del telosoma:
- Mediante procedimientos químicos se ligan los complejos proteicos que se puedan formar incluso por tiempos muy cortos.
- Se purifican en columnas de afinidad en las que se emplean anticuerpos contra TIN2
- Se revierte el ligamiento químico, se digiere mediante endoproteasas y se purifican los péptidos en fase sólida a microescala.
- Se identifican los péptidos mediante un refinamiento de la espectroscopia de masas llamado nano-LC-FTICRMS (del inglés: nano-liquid chromatography- linear quadrupole ion trap-Fourier transformion cyclotron resonance mass spectrometry: Espectrometría de masas de resonancia de ciclotrón con trampa iónical lineal cuadrupolo de transformada de Fourier en fase de nanolíquido).[14]
Componentes y estructura en telosomas de mamífero
Actualmente es el modelo mejor comprendido y estudiado. El telosoma es un complejo multiproteico cuya masa molecular es aproximadamente 1 MDa.[12] Está compuesto por seis proteínas: POT1, TRF1 y TRF2, que se unen directamente al ADN, y TIN2, TPP1 y RAP1, que se unen a las anteriores.[3]
ADN telomérico
Los telosomas se unen con fuerza y especificidad a la secuencia telomérica. Se ha visto que en su posición obliga al ADN a formar una estructura especial que se denomina Bucle-T.[2] Una de las primeras conclusiones que se obtuvieron es la verificación de que todo el ADN telomérico está dentro de esta conformación. Este ADN consiste en secuencias repetitivas no codificantes, (el motivo repetido en humanos en la hebra 3' es [TTAGGG]n), añadidas por una ribonucleoproteína, la telomerasa. Su longitud oscila en humanos entre 2 y 50 kilobases. La hebra 5', al ser complementaria de esta secuencia, es rica en citosina, con lo que habitualmente se la denomina hebra C, mientras que a su complementaria se la denomina hebra T. Ésta última termina en un segmento de unos 75-300 nucleótidos en forma de ADN de cadena sencilla. Este segmento, denominado "saliente" (overhang) invade la doble hebra del telómero en un punto próximo a su comienzo.[11] En 2007 un equipo de científicos de la ENS de Lyon, dirigidos por Eric Gilson, elucidaron la forma en que la proteína telosómica TRF2 cataliza esta invasión. Utilizando plásmidos y observaciones mediante microscopía de fuerza atómica propusieron que la TRF2, acoplándose cerca del saliente 3', obliga al ADN a superenrollarse en sentido dextrógiro, lo que hace que el ADN vecino se desenrolle, favoreciendo de esta manera la invasión de la doble hebra por el saliente 3'.[15] [16] Esta zona en la que el saliente 3' se aparea con su propia hebra, y en la que se pueden encontrar tres hebras de ADN simultáneamente (ADN triplex), se denomina bucle D (D-loop, de Displacement). Una sola repetición telomérica es suficiente para la invasión. Parece ser que la proteína telosómica POT1 se sitúa cubriendo esta zona, y tal vez protegiendo de ese modo el extremo del telómero.[12]
Subunidades proteicas que se unen al ADN
TERF1
La Trf1 humana es una proteína de aproximadamente 60 KDa. Sólo se presenta en forma de homodímero (dos unidades idénticas unidas), incluyendo en el telosoma. Su nombre proviene de las siglas en inglés Telomeric Repeat-Binding Factor 1 (factor de unión a repeticiones teloméricas 1).[17] Su secuencia contiene 439 aminoácidos.[18] El gen está localizado en el cromosoma 8.[17]
Es la proteína más importante del telómero, y la más abundante de las seis del complejo. Se expresa de forma continua a lo largo de todo el ciclo celular, y se une exclusivamente a las repeticiones teloméricas con una fuerza cuatro veces superior a la de su homóloga TRF2. Además de localizarse en el telómero, puede formar estructuras de alto orden con otros dímeros similares sobre el mismo cromosoma, o incluso de otros.[19]
- Estructura
Los rasgos estructurales más destacados de esta proteína son: una estructura con 9 α-hélices y la presencia de un extremo N-terminal rico en aminoácidos con cadena lateral acídica y dos dominios distintos: Uno próximo al extremo C-terminal, tipo c-myb, que es un motivo estructural que también se encuentra en plantas y levaduras y conocido en otras fuentes bibliográficas como "telobox" . Se une específicamente al ADN telomérico.[20] El otro es un motivo central, de unos 200 aminoácidos exclusivo de la familia de proteínas TRF, conocido como dominio de homología TRFH. Habitualmente la proteína dimeriza mediante este dominio, utilizando como interfaz las hélices 1, 2 y 9 de cada monómero.[21]
- Interacciones
Mediante el dominio de homología Trf1 interacciona con TIN2, así como con PINX1.[22] Se ha visto que para que se produzca su unión al ADN telomérico, debe estar fosforilada en la serina-435. Además, para que este punto de fosforilación esté accesible, debe ser fosforilada primero por Cdk1. Parece ser, por tanto, que este evento está regulado por el ciclo celular y es máximo durante la mitosis.[23] También se ha visto que la forma fosforilada podría ser sustrato de Pin1 una peptidil-prolil cis/trans isomerasa esencial para su función. Cuando Pin1 se inhibe, Trf1 se fija demasiado a los telómeros y se distorsiona de esta forma su regulación dinámica, perdiéndose gradualmente los telómeros. En ratones deficientes en esta enzima se produce un envejecimiento prematuro mucho más rápido que en congéneres deficientes en telomerasa.[24]
También se ha visto que se une a las tanquirasas, unas PARPs (poli ADP-ribosa polimerasa), es decir, unas proteínas que transfieren ADP-ribosa procedente del NAD a otras proteínas (PARsilación), cosa que hace con TRF1. Esta modificación inhibe su unión a telómeros. Es notable el hecho de que, al carecer de señal de importación nuclear, debe ser la propia TRF1 la que reclute esta proteína desde su habitual localización citoplasmática, donde se une a otras ocho distintas en localizaciones tan diferentes como la membrana plasmática, el huso mitótico o el aparato de Golgi.[25]
- Función
Las principales funciones son:
- Localización del resto de los componentes: cuando se inactiva TRF1, los demás componentes del telosoma no adquieren una localización correcta. Cuando se carece de la enzima funcional, sus efectos en el fenotipo pueden ser rescatados si se consigue por otros medios que el resto de componentes del telosoma ocupen su lugar. Sin embargo, aún se observan señales teloméricas anormales y elongaciones del telómero anormales. De estos estudios se deduce que su función consiste, de un lado, en el reclutamiento y anclaje del resto de los componentes del telosoma, y parece que existe un papel más directo en el mantenimiento de los telómeros.[26]
- Cohesión de los telómeros en el proceso de mitosis. Parece ser que junto con TIN2, TRF1 se asocia con el complejo de la cohesina durante la mitosis, cumpliendo en los telómeros el mismo papel que estas últimas en los cromosomas.[25]
TERF2
La variante humana de TRF2 es una proteína de 55 534 Da. cuyo punto isoeléctrico es 9,81. El gen que la codifica está localizado en el cromosoma 16 (16q22.1).[27] Su nombre procede de las mismas siglas en inglés que TRF1. Su secuencia consta de 500 aminoácidos y forma un Homodímero (un dímero constituido por dos unidades idénticas). Está presente en dos isoformas.[28] Posee los mismos motivos estructurales que TERF1, pero no puede heterodimerizar con ésta. Además, otra diferencia importante es que su extremo N-terminal es básico.[20]
- Interacciones
TRF2 tiene también un dominio de homología, TRFH, de secuencia distinta, pero con la misma arquitectura molecular.[22] Como en el caso de TRF1, homodimeriza a través de este dominio. Sin embargo, a diferencia de TRF1, no lo utiliza para unirse a TIN2, sino que emplea una región distinta. En lugar de ello, el dominio TRFH de TRF2 se une a una proteína accesoria, Apollo.[29] Se sabe que el daño en el ADN induce la fosforilación de TRF2, aunque de manera transitoria. Al menos una proteína parece tener esta capacidad, la proteína quinasa Aurora C.[30] [31] En este mismo contexto, varias proteínas reparadoras también pueden unirse a TRF2. Ha sido completamente verificado en el caso de WNR,[32] y BLM, aunque en este caso también puede unirse a TRF1. La regulación de la función de esta helicasa podría estar coordinada por ambos dímeros.[33] Así mismo se ha visto que TRF2 puede unirse al complejo de reconocimiento de origen, concretamente a su componente ORC1. Parece ser que esta unión, además de las funciones en el ciclo celular que discutiremos más adelante, cumple un papel en la homeostasis de los telómeros, es decir, en la regulación de su longitud.[34]
Por último, se han observado interacciones con NBN, RAD50, SUB1 y XRCC6.[35]
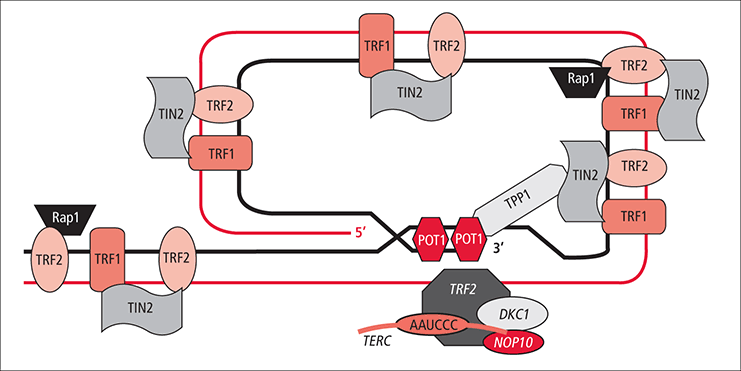
 Diagrama de las principales proteínas o sistemas que interactúan con los componentes del telosoma.
Diagrama de las principales proteínas o sistemas que interactúan con los componentes del telosoma.- Función
Existen diversas evidencias de que tanto TRF1 como TRF2 están implicados en varios procesos epigenéticos en parte dependientes de un efecto de posición, y regulados por el ciclo celular:
- Para comenzar, se puede observar que en queratinocitos transformados en interfase, los telómeros se sitúan en el tercio interior del núcleo celular formando territorios que no se solapan. TERF2 está presente en los telómeros durante todo el ciclo, pero en la transición entre las fases S/G2 aumenta su síntesis, de modo que cuando la célula entra en mitosis, puede localizarse a lo largo de todo el cromosoma, lo que en principio indicaba un papel de esta proteína en la mitosis. Parece ser que tanto la posición de los telómeros como el aumento de síntesis de TERF2 están mediados por la proteína c-myc.[36]
- Una nueva técnica de microscopía confocal que evita el fotoblanqueo, conocida como CLEM, permite la observación durante tiempos prolongados de células vivas, pudiéndose registrar de ese modo la dinámica de algunos procesos durante el ciclo celular. Utilizando esta técnica, un estudio reciente amplió el citado anteriormente comprobando que los telómeros humanos se sitúan en la zona fronteriza entre la eucromatina y la heterocromatina. Los cromosomas acrocéntricos no cumplen exactamente esta regla, situándose más más profundamente en la eucromatina, siempre cerca de unas secuencias de ADN subtelocéntrico repetidas llamadas D4Z4. Es interesante destacar que todos los cromosomas de ratón son acrocéntricos. En esta localización se asocian en "territorios" formados por dos o tres telómeros, con cierta capacidad de movimiento aleatorio y colisiones en las inmediaciones de su posición, que cesa si se elimina el ATP del medio.
- Se ha propuesto que el TRF2 es esencial para la formación de estos territorios, en cooperación con otros componentes recientemente descubiertos. Uno de ellos es el ARN telomérico, que se asocia con un complejo que se ha denominado TERRA. Se observa, como indicio, que la síntesis de TERRA desciende mucho al inicio de la mitosis, y aunque la expresión de TRF2 suele ser bastante constante, queda muy poco cerca de los telómeros, y además, sus propiedades de unión cambian, haciéndose más lábil. Se puede decir lo mismo de TRF1. Se ha atribuido la posición fronteriza de los complejos a un comportamiento epigenético ambiguo y cambiante de los mismos. Se sabe que los telómeros ejercen un efecto de silenciamiento posicional, y son heterocromáticos, pero al mismo tiempo son transcritos en ARN, además de ser vistos muy cerca de dominios nucleares SC35, que son ricos en factores de splicing y permisivos en cuanto a transcripción. Se debe añadir que los telómeros son más abiertos en cuanto a sus efectos epigenéticos cuando se acortan.[37]
- El papel de TRF2 en la regulación epigenética de la cromatina próxima a los telómeros parece aún más importante después del trabajo publicado por el equipo de la investigadora española María Blasco. El ADN próximo a los telómeros muestra habitualmente signos de que sus genes están silenciados, y que por tanto forman parte de la heterocromatina: Las histona 4 está trimetilada en su lisina 20, y hay una intensa metilación de las repeticiones subteloméricas. En ratones que sobreexpresan TRF2, los telómeros se acortan, y desaparecen las señales para la formación de heterocromatina, además de reducirse la presencia de transcritos teloméricos.[38]

Pot1
La isoforma 1 codificada por POT1 es una proteína de 71 KDa perteneciente a la familia de la telombina y contiene 634 aminoácidos. Su nombre proviene de las siglas en inglés "Protection Of Telomeres".[39] El gen de esta proteína en humanos se localiza en el cromosoma 7 (7q31.33). Contiene 22 exones, de manera que da lugar a cinco variantes (isoformas), por splicing alternativo, denominadas por siglas consecutivas v1-v5, estando las cuatro últimas truncadas por su extremo amino o carboxilo. Aún no se sabe su función, pero se especula que las distintas formas pueden cooperar en la protección de telómeros, y en concreto, la forma -COOH truncada v5 se expresa preferencialmente en tumores producidos por un déficit en la reparación de apareamientos de bases erróneos, por lo que parece tener un papel en esta función.[40] Una de estas isoformas parece ser específica de leucocitos.[39] Su mayor nivel de expresión se alcanza en el tejido testicular, mientras que el menor corresponde al músculo esquelético y al colon. Utilizando análisis por PSI-BLAST parece que es la proteína del complejo más conservada en la evolución.[39]
- Estructura
La estructura de POT1 ha sido revelada por cristalografía de rayos X. Para ello se unió a una secuencia de diez nucleótidos de ADN monocatenario telomérico, puesto que se vio que este es el menor número de nucleótidos que necesita para que la unión sea lo suficientemente fuerte. Esto representó una sorpresa, ya que su ortólogo en Schizosaccharomyces pombe se une a seis, de modo que lo esperado es un múltiplo de seis. Se ha visto que contiene dos pliegues de unión a oligonucleótido/oligosacárido (pliegue OB), uniéndose el más próximo al aminoácido N-terminal a los seis primeros nucleótidos, mientras que el segundo se une y protege a otros seis en el saliente 3' de cadena sencilla del telómero. Las ranuras que unen ADN de cada dominio forman un canal continuo entre ambas.[41]
- Uniones
- TPP1 se une a POT1 por su extremo carboxilo. Esta unión es tan constante, y se observa una estequiometría in vivo tan próxima a 1:1, que con frecuencia se habla del "heterodímero POT1-TPP1".[42] [43] Parece que esta interacción es necesaria para la localización de POT1 en telómeros, y así mismo para la regulación de la longitud de los mismos interactuando con la telomerasa conjuntamente.[44] Parece, asimismo, que estas dos proteínas pudieran derivar de una única ancestral, según se desprende del análisis de la proteína de unión a extremos teloméricos de ciliados.[45] Esta unión, además, inhibe la unión de RPA a las porciones de ADN en forma de cadena simple, lo que podría ser una señal errónea de daño.[46] Por último se ha visto que la unión de TPP1 a POT1 aumenta mucho la capacidad de discriminar el ADNss del ARN como sustrato de unión.[47]
- TERF2 se puede unir a POT1. Esta unión no es necesaria para la función de protección de telómeros, pero aumenta la captación de POT1 por estos últimos.[46] [48]
- Función
Fundamentalmente dos:
- Regula la longitud de los telómeros actuando como activador o inhibidor de la telomerasa dependiendo de la posición de POT1 en el saliente 3'. Se le ha comparado a un interruptor de la telomerasa.
- Contribuye a la protección de los telómeros, inhibiendo la formación de puentes anafásicos, y evitando que los sistemas de reparación del ADN reconozcan erróneamente el saliente 3' como daño en el ADN.
Se ha visto que para realizar estas funciones, aumenta la capacidad de las helicasas WRN y BLM de desenrollar el ADN telomérico.[40]
Subunidades proteicas que interconectan el complejo
Tpp1
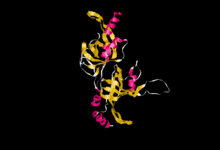 Estructura hallada por cristalografía de rayos X de un péptido que comprende los residuos 90–243 del dominio N-terminal de la proteína TPP1 humana, en la que se puede observar el Pliegue OB. PDB ID: 2I46.
Estructura hallada por cristalografía de rayos X de un péptido que comprende los residuos 90–243 del dominio N-terminal de la proteína TPP1 humana, en la que se puede observar el Pliegue OB. PDB ID: 2I46.Su gen también es conocido como ACD (de Adrenocortical dysplasia homolog).
La Tpp1 humana es una proteína codificada en humanos por el gen ADC en el cromosoma 16q22.1. Tiene 544 aminoácidos y un peso molecular de 57,7 KDa. Posee 2 isoformas, siendo la más larga la primera.[49] [50] Recientemente, el equipo de María Blasco ha probado que esta proteína es indispensable para el reclutamiento de la telomerasa in vivo.[51]
Tin2
El gen que codifica la proteína Tin2 humana, TINF2, consta de nueve exones en una secuencia de 3032 pares de bases, dando lugar a dos transcritos, la isoforma 1, con los nueve exones y 451 aminoácidos y una alternativa de seis exones. Está localizado en el cromosoma 14 (14q11.2).Se desconoce la diferencia funcional de ambas formas.[52] [53]
- Función
- Inhibe la PARsilación de TRF1 formando un complejo ternario con las tanquirasas y regulando de este modo su actividad.[25]
Rap1
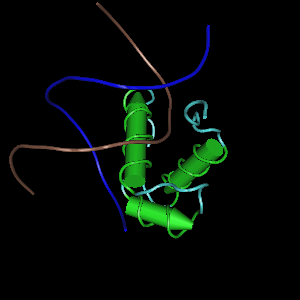
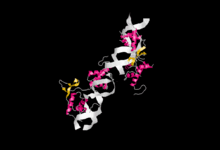 Modelo de cintas de la estructura molecular de RAP1 (en color) junto con ADN (en el centro, en gris).
Modelo de cintas de la estructura molecular de RAP1 (en color) junto con ADN (en el centro, en gris).La proteína Rap1 humana está codificada por el gen TERF2IP, localizado en el cromosoma 16 (16q23.1), con pseudogenes en el cromosoma 5 y 22.[54]
Estequiometría
Diversos estudios han determinado la estequiometría del complejo in vivo. Se ha visto que es similar para células normales o tumorales, y que no se correlaciona con la longitud del telómero:
- Los homodímeros de unión a ADN (TRF1 y TRF2) se encuentran en cantidad suficiente para cubrir por completo los telómeros si son cortos.
- El heterodímero TPP1·POT1 está presente en 50–100 copias/telómero, lo cual excede la longitud de ADN telomérico de cadena simple que debe cubrir. Esto implica que parte de este heterodímero no está asociado al ADN.
- TRF2 y Rap1 se encuentran en proporción 1:1, lo mismo que TPP1 y POT1.
- Hay TIN2 suficiente para que se unan todas las TRF1 y TRF2. TPP1 y POT1 son 10 veces menos abundantes que TIN2.[42]
Evolución
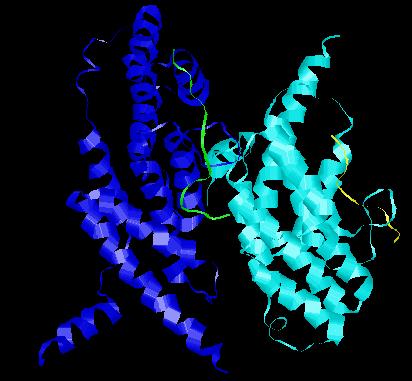
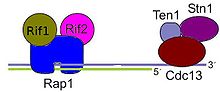 Esquema de las proteínas de unión al final del telómero (complejo CTS) de levadura.
Esquema de las proteínas de unión al final del telómero (complejo CTS) de levadura.En la actualidad se ha visto que los esquemas inicialmente identificados en levaduras y mamíferos están presentes en múltiples Filos, y aunque la arquitectura y la composición de esos complejos varían entre organismos, la función está ampliamente conservada. Las variaciones consisten sobre todo en migraciones o duplicaciones génicas que crean parálogos que asumen nuevas funciones, para la biología del telómero. Esta evolución pudo darse a una velocidad elevada.[55]
- Las primeras proteínas de unión específica al ADN telomérico monocatenario se descubrieron en el protozoo Oxytricha nova, y se conocen como TEBP's (por sus siglas en inglés, Telomere End Binding Proteins).[56] Estas proteínas en organismos sencillos constan de dos subunidades (α y β) formando un complejo ternario con el ADN telomérico. La subunidad α posee dos pliegues OB, mientras que la subunidad β tiene uno más. El contacto principal se efectúa por los dos pliegues N-terminales de la subunidad α y el pliegue OB de la subunidad β formando una amplia hendidura de unión a ADN.[57]
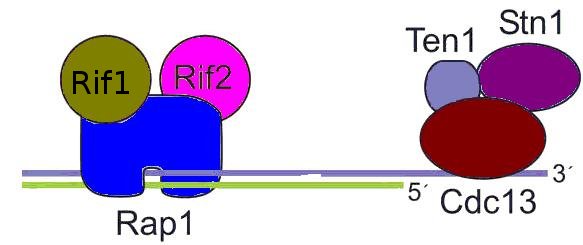
- En Saccharomyces cerevisiae, y en general en hongos el equivalente al telosoma se conoce como Complejo CTS y está formado principalmente por tres proteínas: Stn1, Ten1 y Cdc13 . La secuencia de Cdc13 no muestra una aparente homología con las proteínas con pliegues OB, los estudios realizados mediante RMN revelaron que existía una conformación similar al pliegue OB de la TEBPα. Las otras dos son más similares a Rpa.[58]
- Aunque en principio se creía que el complejo CTS y el basado en POT1 eran incompatibles, existen proteínas asociadas a telómeros en S. cerevisiae similares a la Shelterina, estas son Rif1, Rif2 y Rap1. Aparentemente realizan las mismas funciones de inhibir la fusión de telómeros por NHEJ y la elongación por la telomerasa, así como el procesamiento nucleolítico de los telómeros en las fases del ciclo celular G1 y G2, es decir, la formación del saliente 3'.[59]
En estos organismos sencillos, el ADN telomérico parece adoptar una conformación de orquilla en lugar de la conformación en T-loop que se aprecia en vertebrados y en plantas.[55] [60]
- En plantas, POT1 suele tener una función distinta, siendo un regulador positivo de la telomerasa.
- Los estudios funcionales y estructurales parecen establecer que la POT1 humana es homóloga de la subunidad α de TEBP de O. nova, mientras que la TPP1 parecería homóloga de TEBPβ, aunque esta última subunidad no se ha encontrado en ningún otro organismo, salvo en el ciliado Stylonychia mytilis, y parece ser que las propiedades de unión del heterodímero POT1–TPP1 al ADN recuerdan a las del TEBPα–β de O. nova.[43]
 El telosoma durante el ciclo celular. Arriba, en las fases de crecimiento (G1 y G2) y mitosis) el complejo está ensamblado, y POT1 protege el saliente 3', al mismo tiempo que se inhibe la acción de la telomerasa y se evita que las enzimas de reparación del ADN reconozcan erróneamente este saliente como una ruptura cromosómica. En la fase de síntesis (fase S) se desacopla el subcomplejo de Pot1 y se favorece la progresividad de la telomerasa, mientras se sigue evitando la acción de las enzimas de reparación del ADN.
El telosoma durante el ciclo celular. Arriba, en las fases de crecimiento (G1 y G2) y mitosis) el complejo está ensamblado, y POT1 protege el saliente 3', al mismo tiempo que se inhibe la acción de la telomerasa y se evita que las enzimas de reparación del ADN reconozcan erróneamente este saliente como una ruptura cromosómica. En la fase de síntesis (fase S) se desacopla el subcomplejo de Pot1 y se favorece la progresividad de la telomerasa, mientras se sigue evitando la acción de las enzimas de reparación del ADN.Patología molecular
Diversas alteraciones de este complejo están relacionadas con afecciones como la disqueratosis congénita, y en muy pocos casos de anemia aplásica, en este caso prácticamente todos ellos pediátricos.[61] [62] [63] Así mismo se especula su posible relación con algunos tipos de cáncer, en la aparición de las enfermedades del envejecimiento y en el propio proceso de envejecimiento.[64]
TINF2 es el primer gen de proteína de la shelterina en el que se demostró que su alteración producía enfermedades genéticas autosómicas dominantes. Se encuentra alterado en aproximadamente el 10% de los casos de disqueratosis congénita.[65] Hoy en día se conocen más de 18 mutaciones puntuales, junto con alguna inserción o deleción, que producen enfermedades como la disqueratosis congénita, y sus variedades (síndrome de Hoyeraal-Hreidarsson, con aplasia cerebelar y síndrome de Revesz, con retinopatía bilateral exudativa y calcificaciones intracraneales) y también aplasia medular. Todas las mutaciones se sitúan en el exón 6a, entre los aminoácidos 236 (prolina) 298 (glutamina), que están próximas o en la misma zona del dominio de unión a Trf1.[66] [22] Además de la disqueratosis y anomalías en la pigmentación, se suelen suceder complicaciones como fallo medular o enfermedades malignas de la médula ósea. En el diagnóstico mediante FISH se observa siempre un acortamiento anormal de los telómeros.[52] [53] El mecanismo patogénico por el que las mutaciones de TINF2 producen la enfermedad no está claro, pero existen algunas hipótesis que establecen que se produce es un impedimento del acceso de la telomeras al final del telómero: Los niveles de ARN telomérico no están elevados en los pacientes, y por tanto probablemente tampoco la actividad de la telomerasa. Sus telómeros también son extraordinariamente cortos. Esta hipótesis además explica las similitudes fenotípicas con los otros genes cuyas mutaciones producen la enfermedad: (DKC1, TERC, TERT, NOP10, NHP2).[65]
Se ha visto que las proteínas de la shelterina están implicadas en la transición de las células de Hodgkin a células de Reed–Sternberg.[67] Las últimas, que son un hallazgo indicativo del linfoma de Hodgkin, proceden de la fusión de las primeras, observándose un gran acortamiento de telómeros en el proceso.[68]
Notas
- ↑ Sunil Kaul y Renu Wadhwa (2003). Aging of Cells in and Outside the Body. springer. ISBN 1-4020-1375-2.
- ↑ a b c d de Lange, T (2005). «Shelterin: the protein complex that shapes and safeguards human telomeres». Genes & Development (19). doi 10.1101/gad.1346005. http://genesdev.cshlp.org/content/19/18/2100.
- ↑ a b Zhou Songyang (2003). «Página personal del Profesor Zhou Songyang en el Baylor College of Medicine» (en inglés). Consultado el 19 de noviembre de 2008.
- ↑ Zellinger, B y Riha, K (2007). «Composition of plant telomeres». Biochimica et Biophysica acta (1769).
- ↑ Rudolph, KL (2008). Telomeres and Telomerase in Aging, Disease, and Cancer: Molecular Mechanisms of Adult Stem Cell Ageing. Springer. ISBN 3-540-73708-1.
- ↑ Robert G. Fenton y Dan L. Longo. Harrison's Principles of Internal Medicine, Cap. 69, pg. 454: Cancer cell biology and angiogenesis.
- ↑ McKnight, TD y Shippen, DE (2004). «Plant telomere biology». The Plant Cell 16. http://www.plantcell.org/cgi/reprint/16/4/794.
- ↑ L. T. Evans, William James Peacock, Otto Herzberg Frankel (1981). Wheat Science, Today and Tomorrow. CUP Archive. ISBN 0-521-23793-9.
- ↑ Gottschling DE y Cech TR (1984). «Chromatin structure of the molecular ends of Oxytricha macronuclear DNA: phased nucleosomes and a telomeric complex». Cell 38 (2). PMID 6432344.
- ↑ a b Wright JH, Gottschling DE, Zakian VA (1992). «Saccharomyces telomeres assume a non-nucleosomal chromatin structure». Genes & development 6 (2). PMID 1737616.
- ↑ a b ack D. Griffith, Laurey Comeau, Soraya Rosenfield, Rachel M. Stansel, Alessandro Bianchi, Heidi Moss y Titia de Lange (1999). «Mammalian Telomeres End in a Large Duplex Loop». Cell 97 (4). http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6WSN-4194R53-C&_user=10&_rdoc=1&_fmt=&_orig=search&_sort=d&view=c&_acct=C000050221&_version=1&_urlVersion=0&_userid=10&md5=6d2efe3cf27df704820661de6ce6c8d1.
- ↑ a b c d Liu D, O'Connor MS, Qin J, Songyang Z. (2004). «Telosome, a mammalian telomere-associated complex formed by multiple telomeric proteins.». J. Biol. Chem 279 (49). PMID 15383534. http://www.jbc.org/cgi/reprint/279/49/51338.
- ↑ Xin H, Liu D, Songyang Z. (2008). «The telosome/shelterin complex and its functions». Genome Biol. 9 (9). PMID 18828880.
- ↑ Nittis, Thalia; Lionel Guittat, Richard D Leduc, Ben Dao, Julien P Duxin, Henry Rohrs, R Reid Townsend, Sheila A Stewart (22-01-2010). «Revealing novel telomere proteins using in vivo crosslinking,tandem affinity purification and label-free quantitative LC-FTICR-MS». Molecular & Cellular Proteomics: MCP. doi:. ISSN 1535-9484.
- ↑ «S’il te plaît, dessine-moi un télomère» (en francés). CNRS. Consultado el 30 de noviembre de 2008.
- ↑ Amiard S, Doudeau M, Pinte S, Poulet A, Lenain C, Faivre-Moskalenko C, Angelov D, Hug N, Vindigni A, Bouvet P, Paoletti J, Gilson E, Giraud-Panis MJ. (2007). «[PMID 17220898 A topological mechanism for TRF2-enhanced strand invasion]». Nat.. Struct. Mol Biol. 14 (2). PMID 17220898. PMID 17220898.
- ↑ a b Broccoli D, Chong L, Oelmann S, Fernald AA, Marziliano N, van Steensel B, Kipling D, Le Beau MM, de Lange T. (1997). «Comparison of the human and mouse genes encoding the telomeric protein, TRF1: chromosomal localization, expression and conserved protein domains». Hum Mol Genet. 6 (1). PMID 9002672.
- ↑ Chong, L.; van Steensel, B.; Broccoli, D.; Erdjument-Bromage, H.; Hanish, J.; Tempst, P.; de Lange, T. (1995). «Comparison of the human and mouse genes encoding the telomeric protein, TRF1: chromosomal localization, expression and conserved protein domains». science 270. PMID 7502076.
- ↑ Matulić M, Sopta M, Rubelj I (2007). «Telomere dynamics: the means to an end». Cell prolif. 40 (4). PMID 17635515.
- ↑ a b T Bilaud, CE Koering, E Binet-Brasselet, K Ancelin, A Pollice, SM Gasser and E Gilson (PMID 8614633). «The telobox, a Myb-related telomeric DNA binding motif found in proteins from yeast, plants and human». Nucleic Acids Research 24 (7). http://nar.oxfordjournals.org/cgi/content/full/24/7/1294.
- ↑ Fairall L, Chapman L, Moss H, de Lange T, Rhodes D. (2001). «Structure of the TRFH Dimerization Domain of the Human Telomeric Proteins TRF1 and TRF2». Mol. Cell 8 (2). PMID 11545737. http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6WSR-4C5RF70-F&_user=10&_rdoc=1&_fmt=&_orig=search&_sort=d&view=c&_acct=C000050221&_version=1&_urlVersion=0&_userid=10&md5=fa8f96164a2b0172b5e1fb0bb901fb86.
- ↑ a b c Chen Y, Yang Y, van Overbeek M, Donigian JR, Baciu P, de Lange T, Lei M. (2008). «A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins». Science 319 (5866). PMID 18202258.
- ↑ Wu ZQ, Yang X, Weber G, Liu X. (Plk1 phosphorylation of TRF1 is essential for its binding to telomeres.). «2008». J. Biol. Chem 283 (37). PMID 18625707.
- ↑ Lee TH, Tun-Kyi A, Shi R, Lim J, Soohoo C, Finn G, Balastik M, Pastorino L, Wulf G, Zhou XZ, Lu KP (2009). «Essential role of Pin1 in the regulation of TRF1 stability and telomere maintenance». Nature 11 (1). PMID 19060891.
- ↑ a b c Hsiao SJ, Smith S. (2008). «Tankyrase function at telomeres, spindle poles, and beyond». Biochemie 90 (1). PMID 17825467.
- ↑ Okamoto K, Iwano T, Tachibana M, Shinkai Y (2008). «Distinct roles of TRF1 in the regulation of telomere structure and lengthening». J. Biol. Chem. 35. PMID 18587156.
- ↑ «Sección sobre TERF2» (en inglés). ``municipio de Piscataway: Gene Script.. Consultado el 8 de diciembre de 2008.
- ↑ «Uniprot Q15554» (en inglés). UniProt. Consultado el 8 de diciembre de 2008.
- ↑ Chen Y, Yang Y, van Overbeek M, Donigian JR, Baciu P, de Lange T, Lei M (2008). «A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins». Science 319 (5866). PMID 18202258.
- ↑ Tanaka H, Mendonca MS, Bradshaw PS, Hoelz DJ, Malkas LH, Meyn MS, Gilley D (2005). «DNA damage-induced phosphorylation of the human telomere-associated protein TRF2». PNAS 102 (43). PMID 16223874. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=16223874.
- ↑ Spengler, D (2007). «The protein kinase Aurora C phosphorylates TRF2». Cell Cycle 6 (20). PMID 18309533.
- ↑ Li B, Jog SP, Reddy S, Comai L. (2008). «WRN controls formation of extrachromosomal telomeric circles and is required for TRF2DeltaB-mediated telomere shortening». Mol. Cell Biol. 28 (6). PMID 18212065. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=18212065.
- ↑ Lillard-Wetherell K, Machwe A, Langland GT, Combs KA, Behbehani GK, Schonberg SA, German J, Turchi JJ, Orren DK, Groden J. (2004). «Association and regulation of the BLM helicase by the telomere proteins TRF1 and TRF2». Hum. Mol. Genet. 13 (17). PMID.
- ↑ Tatsumi Y, Ezura K, Yoshida K, Yugawa T, Narisawa-Saito M, Kiyono T, Ohta S, Obuse C, Fujita M. (2008). «Involvement of human ORC and TRF2 in pre-replication complex assembly at telomeres». Genes cells. 13 (10). PMID 18761675.
- ↑ «TERF2» (en inglés). AceView-NCBIy la NLM. Consultado el 12 de enero de 2009.
- ↑ Ermler S, Krunic D, Knoch TA, Moshir S, Mai S, Greulich-Bode KM, Boukamp P (2004). «Cell cycle-dependent 3D distribution of telomeres and telomere repeat-binding factor 2 (TRF2) in HaCaT and HaCaT-myc cells». Eur. J. Cell Biol. 83 (11-12). PMID 15679112.
- ↑ De Vos WH, Hoebe RA, Joss GH, Haffmans W, Baatout S, Van Oostveldt P, Manders EM (2008). «Controlled light exposure microscopy reveals dynamic telomere microterritories throughout the cell cycle». Citometry A.. PMID 19097172.
- ↑ Benetti R, Schoeftner S, Muñoz P, Blasco MA (2008). «Role of TRF2 in the assembly of telomeric chromatin». Cell Cycle 7 (21). PMID 18971622.
- ↑ a b c Baumann P, Podell E, Cech TR (2002). «Human Pot1 (protection of telomeres) protein: cytolocalization, gene structure, and alternative splicing.». Mol. Cell. Biol. 22 (22): pp. 8079–87. PMID 12391173.
- ↑ a b Yang Q, Zhang R, Horikawa I, Fujita K, Afshar Y, Kokko A, Laiho P, Aaltonen LA, Harris CC (2007). «Functional diversity of human protection of telomeres 1 isoforms in telomere protection and cellular senescence». Cancer Res 67 (24). PMID 18089797. http://cancerres.aacrjournals.org/cgi/content/full/67/24/11677.
- ↑ Lei M, Podell ER, Cech TR (2005). «Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection.». Nat. Struct. Mol. Biol. 11 (12): pp. 1223–9. doi:. PMID 15558049.
- ↑ a b Kaori K. Takai,Sarah Hooper1,Stephanie Blackwood2,Rita Gandhi3 y Titia de Lange (enero de 2010). «In Vivo Stoichiometry of Shelterin Components». Journal of Biological Chemistry, (285): pp. 1457-1467.. doi:.
- ↑ a b Wang, Feng; Elaine R. Podell, Arthur J. Zaug, Yuting Yang, Paul Baciu, Thomas R. Cech, Ming Lei (2007). «The POT1–TPP1 telomere complex is a telomerase processivity factor». Nature 445 (7127): pp. 506-510. doi:. ISSN 0028-0836.
- ↑ Liu D, Safari A, O'Connor MS, Chan DW, Laegeler A, Qin J y Songyang Z (2004). «PTOP interacts with POT1 and regulates its localization to telomeres.». Nat Cell Biol 6 (7). PMID 15181449.
- ↑ Xin H, Liu D, Wan M, Safari A, Kim H, Sun W, O'Connor MS, Songyang Z (2007). «TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase». Nature 445 (7127). PMID 17237767.
- ↑ a b Barrientos KS, Kendellen MF, Freibaum BD, Armbruster BN, Etheridge KT, Counter CM. (2008). «Distinct functions of POT1 at telomeres.». Mol Cell Biol. 17. PMID 18519588. http://mcb.asm.org/cgi/content/full/28/17/5251?view=long&pmid=18519588.
- ↑ Nandakumar, Jayakrishnan; Elaine R Podell, Thomas R Cech (12-01-2010). «How telomeric protein POT1 avoids RNA to achieve specificity for single-stranded DNA». Proceedings of the National Academy of Sciences of the United States of America 107 (2): pp. 651-656. doi:. ISSN 1091-6490.
- ↑ Yang Q, Zheng YL, Harris CC. (2005). «POT1 and TRF2 cooperate to maintain telomeric integrity.». Mol. Cell. Biol. 25 (3). PMID 15657433. http://mcb.asm.org/cgi/content/full/25/3/1070?ijkey=65c81d14f71eb363e8bfd2e8252a51c0a41c1a5f.
- ↑ «ACD adrenocortical dysplasia homolog (mouse) [ Homo sapiens ]». NCBI Entrez Gene. Consultado el 26 de mayo de 2010.
- ↑ «adrenocortical dysplasia homolog (mouse)». Consultado el 1 de junio de 2010.
- ↑ «TPP1 is required for TERT recruitment, telomere elongation during nuclear reprogramming, and normal skin development in mice». Dev Cell 18 (5): pp. 691-2. mayo de 2010. PMID 20493811.
- ↑ a b «Telomerase database-diseases/TINF2». Consultado el 7 de junio de 2010.
- ↑ a b Sharon A Savage (noviembre de 2009). NCBI GeneReviews (ed.): «Dyskeratosis congenita:Molecular Genetics». Consultado el 7 de junio de 2010.
- ↑ «Entrez Gene 54386». Consultado el 29 de marzo de 2011.
- ↑ a b Linger, Benjamin R.; Carolyn M. Price (2009). «Conservation of telomere protein complexes: shuffling through evolution». Critical Reviews in Biochemistry and Molecular Biology 44 (6): pp. 434-446. doi:. ISSN 1040-9238.
- ↑ Gottschling, D (1986). «Telomere proteins: Specific recognition and protection of the natural termini of Oxytricha macronuclear DNA». Cell 47 (2): pp. 195-205. doi:. ISSN 00928674.">
- ↑ Gottschling, D (1986). «Telomere proteins: Specific recognition and protection of the natural termini of Oxytricha macronuclear DNA». Cell 47 (2): pp. 195-205. doi:. ISSN 00928674.
- ↑ Baumann, Peter; Carolyn Price (21-05-2010). «Pot1 and telomere maintenance». FEBS Letters. doi:. ISSN 1873-3468.
- ↑ Bonetti, Diego; Michela Clerici, Savani Anbalagan, Marina Martina, Giovanna Lucchini, Maria Pia Longhese, James E. Haber (ed.) (2010). «Shelterin-Like Proteins and Yku Inhibit Nucleolytic Processing of Saccharomyces cerevisiae Telomeres». PLoS Genetics 6 (5): pp. e1000966. doi:. ISSN 1553-7404.
- ↑ Shakirov, E. V.; X. Song, J. A. Joseph, D. E. Shippen (2009). «POT1 proteins in green algae and land plants: DNA-binding properties and evidence of co-evolution with telomeric DNA». Nucleic Acids Research 37 (22): pp. 7455-7467. doi:. ISSN 0305-1048.
- ↑ Rook, Arthur (2010). Rook's textbook of dermatology. (8th ed. edición). Chichester West Sussex UK ;Hoboken NJ: Wiley-Blackwell. ISBN 9781405161695.
- ↑ Du, Hong-Yan; Philip J. Mason, Monica Bessler, David B. Wilson (2009). «TINF2mutations in children with severe aplastic anemia». Pediatric Blood & Cancer 52 (5): pp. 687-687. doi:. ISSN 15455009.
- ↑ Calado, R. T.; N. S. Young (2008). «Telomere maintenance and human bone marrow failure». Blood 111 (9): pp. 4446-4455. doi:. ISSN 0006-4971.
- ↑ Oeseburg, Hisko; Rudolf A. Boer, Wiek H. Gilst, Pim Harst (2009). «Telomere biology in healthy aging and disease». Pflügers Archiv - European Journal of Physiology 459 (2): pp. 259-268. doi:. ISSN 0031-6768.
- ↑ a b Bessler, Monica; David B. Wilson, Philip J. Mason (2010). «Dyskeratosis congenita». FEBS Letters. doi:. ISSN 00145793.
- ↑ OMIM 604319
- ↑ Knecht, H; B Sawan, D Lichtensztejn, B Lemieux, R J Wellinger, S Mai (2008). «The 3D nuclear organization of telomeres marks the transition from Hodgkin to Reed–Sternberg cells». Leukemia 23 (3): pp. 565-573. doi:. ISSN 0887-6924.
- ↑ Riede, Ursus-Nikolaus (2004). Color atlas of pathology : pathologic principles, associated diseases, sequela. Stuttgart ;;New York: Thieme. ISBN 9783131277817.
Categorías:- Genética molecular
- Cromosomas
- Complejos proteicos


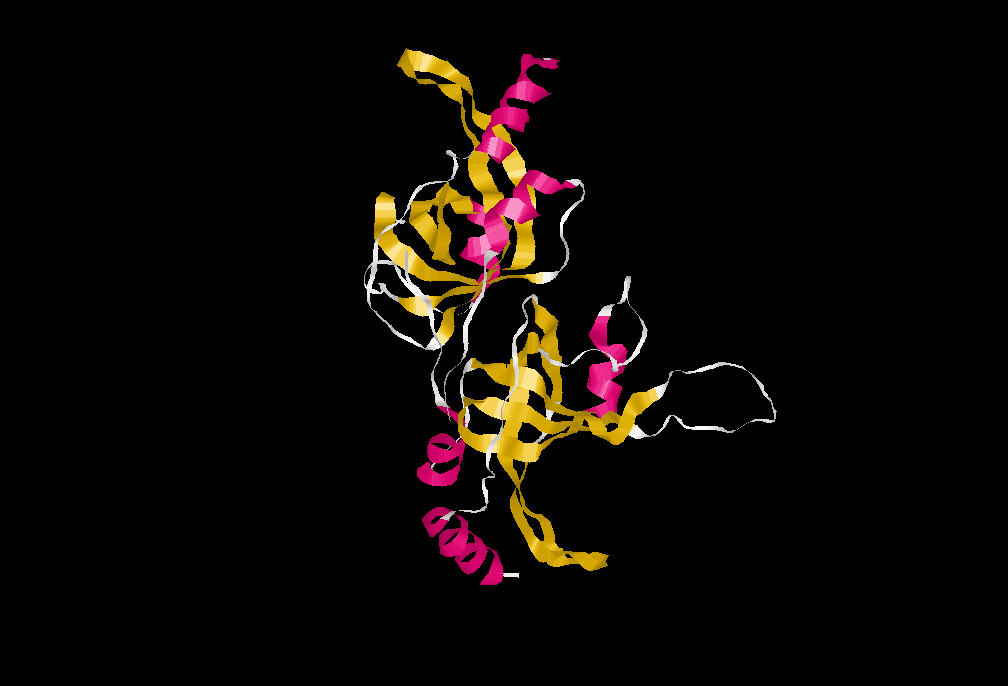
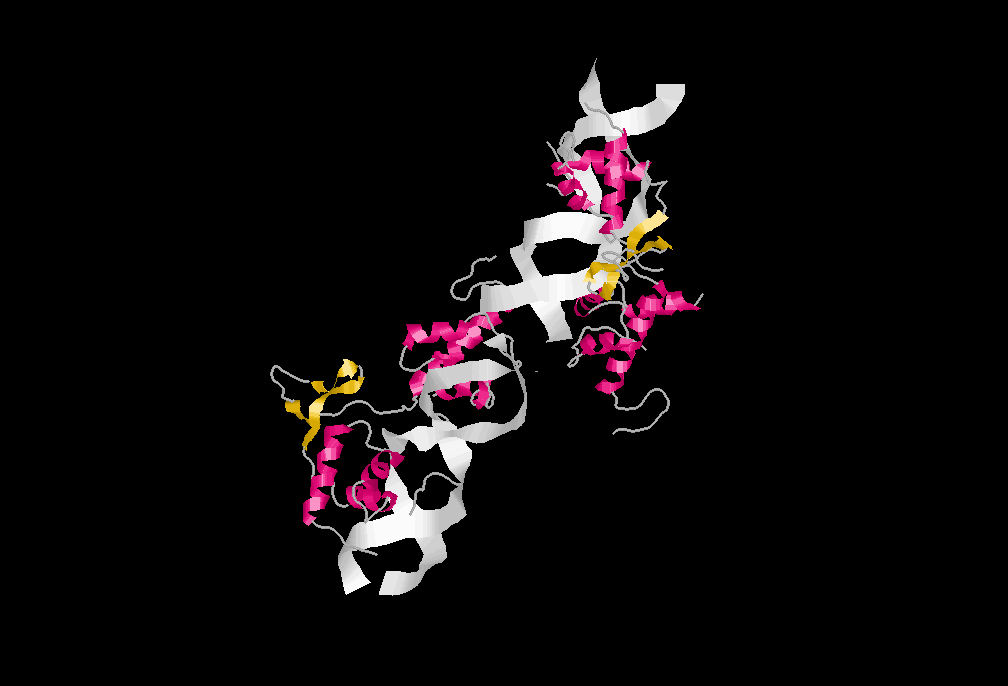

Wikimedia foundation. 2010.