- Paracetamol
-
Paracetamol 
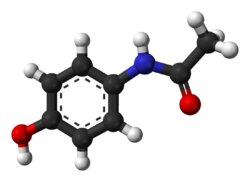
Nombre (IUPAC) sistemático N-(4-hidroxifenil)etanamida Identificadores Número CAS 103-90-2 Código ATC N02BE01 PubChem 1983 DrugBank DB00316 ChemSpider 1906 UNII 362O9ITL9D KEGG D00217 ChEBI 116450 Datos químicos Fórmula C8H9NO2 Peso mol. 151.17 g/mol SMILESCC(=O)Nc1ccc(cc1)OInChI=1S/C8H9NO2/c1-6(10)9-7-2-4-8(11)5-3-7/h2-5,11H,1H3,(H,9,10)
Key:RZVAJINKPMORJF-UHFFFAOYSA-NDatos físicos Densidad 1,263 g/cm³ P. fusión 169 °C (336 °F) Solubilidad en agua 12,78 mg/mL (20 °C) Farmacocinética Biodisponibilidad ~100% Metabolismo 90 to 95% Hepático Vida media 1–4 h Excreción Renal Datos clínicos Inf. de Licencia FDA:enlace Cat. embarazo A (AU) B (USA) Seguro Estado legal Unscheduled (AU) GSL (UK) OTC (USA) Vías de adm. Oral, Rectal, Intravenosa  Aviso médico
Aviso médico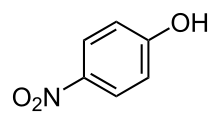 4-nitrofenol, precursor de la síntesis de p-acetaminofeno
4-nitrofenol, precursor de la síntesis de p-acetaminofeno
El paracetamol (DCI) o acetaminofén (acetaminofeno) es un fármaco con propiedades analgésicas, sin propiedades antiinflamatorias clínicamente significativas. Actúa inhibiendo la síntesis de prostaglandinas, mediadores celulares responsables de la aparición del dolor. Además, tiene efectos antipiréticos. Se presenta habitualmente en forma de cápsulas, comprimidos, supositorios o gotas de administración oral.
Es un ingrediente frecuente de una serie de productos contra el resfriado común y la gripe. A dosis estándar es casi seguro, pero su bajo precio y amplia disponibilidad han dado como resultado frecuentes casos de sobredosificación. En las dosis indicadas el paracetamol no afecta a la mucosa gástrica ni a la coagulación sanguínea o los riñones, pero sí al hígado, severamente (véase:Toxicidad).
A diferencia de los analgésicos opioides, no provoca euforia ni altera el estado de humor del paciente. Al igual que los antiinflamatorios no esteroideos (AINEs), no se asocia con problemas de adicción, tolerancia y síndrome de abstinencia.
Los nombres paracetamol y acetaminofén pertenecen a la historia de este compuesto y provienen de la nomenclatura tradicional de la química orgánica, N-acetil-para-aminofenol y para-acetil-aminofenol.
Según las recomendaciones de 1993 de la IUPAC, el nombre de este compuesto es N-(4-hidroxifenil)etanamida.
Contenido
Historia
En la antigüedad y durante el Medievo, los únicos agentes antipiréticos conocidos eran compuestos presentes en la corteza del sauce (una familia de compuestos conocidos como salicilinas, los cuales finalmente dieron lugar al ácido acetilsalicílico), y otros contenidos en la corteza de la quina. La corteza de la quina asimismo era usada para la obtención de quinina, compuesto con actividad antimalaria. La quinina en sí misma también tiene actividad antipirética. Los esfuerzos para aislar y purificar la salicilina y el ácido salicílico tuvieron lugar durante mediados y finales del siglo XIX.
Cuando la quina empezó a escasear en los años 1880, la gente empezó a buscar alternativas. Dos agentes antipiréticos alternativos fueron desarrollados en los años 1880: la acetanilida en 1886 y la fenacetina en 1887. En ese momento, el paracetamol ya había sido sintetizado por Harmon Morse de Northrop mediante la reducción del p-nitrofenol en ácido acético glacial. Este hecho se produjo en 1873, y el paracetamol no se usó con fines médicos durante dos décadas. En 1893, el paracetamol fue encontrado en la orina de personas que habían ingerido fenacetina y fue aislado como un compuesto blanco y cristalino de sabor amargo. En 1899, el paracetamol fue identificado como un metabolito de la acetanilida. Dicho descubrimiento fue ampliamente ignorado en aquel momento.
 Julius Axelrod, galardonado con el Premio Nobel en 1970
Julius Axelrod, galardonado con el Premio Nobel en 1970En 1946, el Instituto para el Estudio de Drogas Analgésicas y Sedantes otorgó una subvención al Ministerio de Sanidad de Nueva York para estudiar los problemas asociados con el uso de analgésicos. Bernard Brodie y Julius Axelrod fueron asignados para investigar por qué compuestos no relacionados con la aspirina daban lugar a metahemoglobinemia, un síndrome no letal consistente en la deformación de la molécula de la hemoglobina y por tanto causante de su incapacidad para transportar oxígeno de forma efectiva. En 1948 ambos investigadores relacionaron el uso de la acetanilida con la metahemoglobinemia y dedujeron que su efecto analgésico era debido a su metabolito paracetamol. Propusieron el uso de paracetamol (acetaminofén) ya que éste no tenía los efectos tóxicos de la acetanilida.[1]
El paracetamol fue puesto a la venta en los Estados Unidos en 1955 bajo el nombre comercial Tylenol. En 1956, pastillas de 500 mg de paracetamol se pusieron a la venta en el Reino Unido bajo el nombre de Panadol, producido por Frederick Stearns & Co, una filial de Sterling Drug Inc. Al principio Panadol estuvo disponible únicamente con receta médica, para el alivio del dolor y la fiebre, y fue anunciado como "inocuo para el estómago", debido a que otros analgésicos de la época contenían ácido acetilsalicílico, un irritante conocido del estómago, pero ya en abril del 2009 la Administración de Alimentos y Medicamentos de EUA obliga a fabricantes a informar que el paracetamol es altamente tóxico, potencialmente mortal, en virtud de los daños que puede causar al hígado.[2] En junio de 1958 se comercializó una formulación para niños, Panadol Elixir. En 1963 el paracetamol se añadió al vademécum británico, y desde entonces se ha popularizado como un analgésico con pocos efectos secundarios y con pocas interacciones con otros medicamentos.
La patente sobre el paracetamol ha expirado en los Estados Unidos, y varios genéricos están ampliamente disponibles bajo el Acta de Competitividad de Precios y la Ley de Restauración de Patentes de 1984, aunque ciertos preparados de Tylenol estuvieron protegidos hasta el 2007. En los EE.UU., la patente número 6.126.967 del 3 de septiembre de 1998 fue concedida para la "liberación extendida de preparados de acetaminofén".
Disponibilidad comercial
 Supositorios de paracetamol
Supositorios de paracetamolNormalmente se comercializa como suspensión líquida, en comprimidos o como supositorio. Dada su amplia disponibilidad, en muchas ocasiones su eficacia está infravalorada.[cita requerida]
El Panadol, que se vende en Europa, América Latina, Asia y Australia, es la marca más extendida, vendida en más de 80 países. En Norteamérica, el paracetamol se vende como genérico o bajo varias marcas: por ejemplo Tylenol (McNeil - PPC, Inc), Anacin 3 y Datril.
En algunas formulaciones el paracetamol se combina con el opioide codeína, a veces llamado co-codamol, como por ejemplo el Algidol (Almirall) en España, lo que lo convierte en más tóxico[cita requerida]. En los Estados Unidos y Canadá se vende como Tylenol 1/2/3/4; en los EE.UU. sólo se puede adquirir con receta médica, lo contrario que en Canadá. En el Reino Unido y en otros muchos países, esta combinación se vende como Tylex CD y Panadeine. Otras marcas disponibles son: Captin, Disprol, Dymadon, Fensum, Hedex, Mexalen, Nofedol, Pediapirin,atamel y Perfalgan. En España puede adquirirse indistintamente como genérico o como medicamento "de marca", como el muy conocido Efferalgan de (Upsa Laboratoires), Termalgin (Novartis), Gelocatil o Frenadol (compuesto con Dextrometorfano, ácido ascórbico y clorfenamina comercializado por McNeil).
También se puede combinar con oxicodona, por ejemplo el Percocet en EEUU.
Síntesis
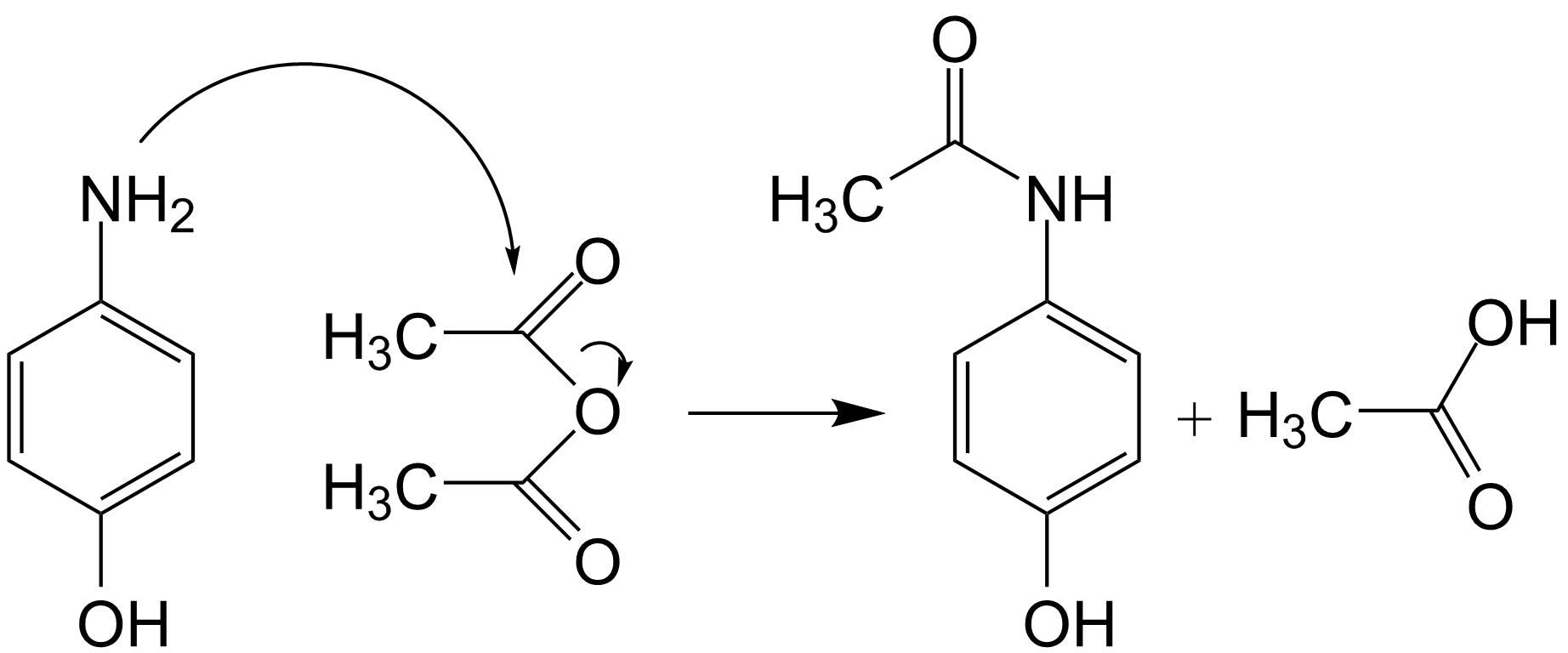
Las reacción del p-aminofenol con anhídrido acético, produce la acetilación del primero, obteniéndose como productos el paracetamol y ácido acético.
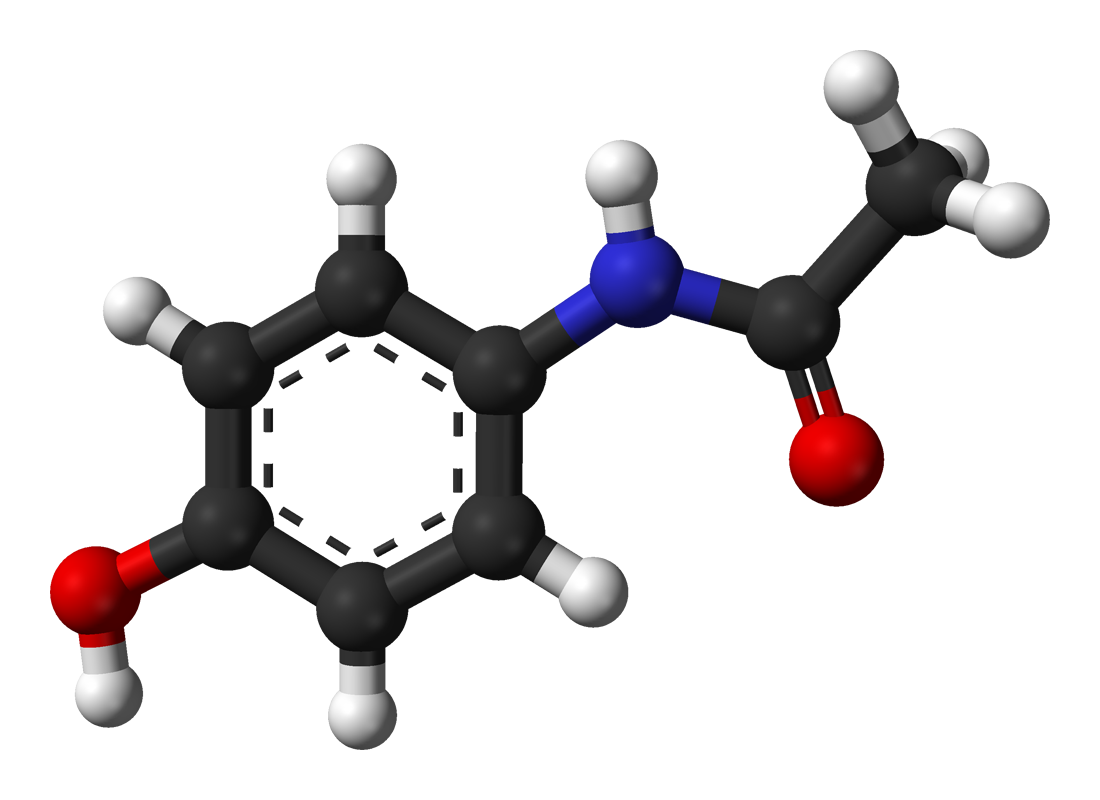
 Síntesis química del paracetamol
Síntesis química del paracetamolAunque conviene proteger el grupo hidroxilo fenólico debido a su mayor poder nucleófilo, respecto al nitrógeno anilínico.
Farmacodinamia
Durante mucho tiempo se ha creído que el mecanismo de acción del paracetamol es similar al del ácido acetilsalicílico (AAS). Es decir, que actúa reduciendo la síntesis de prostaglandinas, compuestos relacionados con los procesos febriles y el dolor, inhibiendo la ciclooxigenasa (COX).
Sin embargo, hay diferencias importantes entre los efectos del ácido acetilsalicílico y el paracetamol. Las prostaglandinas participan en los procesos inflamatorios, pero el paracetamol no presenta actividad antiinflamatoria apreciable. Además, la COX también participa en la síntesis de tromboxanos que favorecen la coagulación de la sangre; el AAS tiene efectos anticoagulantes, pero el paracetamol no. Finalmente, el AAS y otros AINEs son perjudiciales para la mucosa gástrica, donde las prostaglandinas desempeñan un papel protector, pero en este caso el paracetamol es seguro.
De esta forma, mientras el AAS actúa como un inhibidor irreversible de la COX y bloquea el centro activo de la enzima directamente, el paracetamol la bloquea indirectamente y este bloqueo es inútil en presencia de peróxidos.[3] Esto podría explicar por qué el paracetamol es eficaz en el sistema nervioso central y en células endoteliales, pero no en plaquetas y células del sistema inmunitario, las cuales tienen niveles altos de peróxidos.
Swierkosz et al. (2002) encontró evidencias que indican que el paracetamol inhibe una variante de la enzima COX que es diferente a las variantes COX-1 y COX-2, denominada ahora COX-3.[4] Su mecanismo de acción exacto no es bien comprendido aún, pero futuras investigaciones pueden esclarecerlo.
Farmacocinética
El paracetamol se absorbe rápida y completamente por vía oral, y bastante bien por vía rectal, teniendo la ventaja de evitar el primer paso hepático. Existen también preparaciones intravenosas. Las concentraciones plasmáticas máximas se alcanzan en función de la forma farmacéutica, con un tiempo, hasta la concentración máxima, de 0,5-2 horas. El paracetamol se distribuye rápidamente por todos los tejidos. Las concentraciones son similares en la sangre, la saliva y el plasma. La tasa de unión a las proteínas plasmáticas es baja. La biodisponibilidad es muy elevada (cercana al 100%),siendo la biodisponibilidad por vía oral del 75-85%. El paracetamol se metaboliza principalmente a nivel del hígado. Las dos principales rutas metabólicas son la glucuro y sulfuroconjugación. Esta última vía se satura rápidamente con dosis superiores a las terapéuticas. Solamente una pequeña proporción se metaboliza mediante el sistema enzimático del citocromo P-450 en el hígado, por acción de las oxidasas mixtas,generando un intermedio reactivo, N-acetilbenzoquinoneimida que en condiciones normales es inactivado (se detoxifica) por reacción con los grupos sulfhidrilo del glutatión y eliminado en la orina conjugado con cisteína y ácido mercaptúrico. Por el contrario, durante las intoxicaciones graves aumenta la cantidad de este metabolito tóxico. Dosis elevadas de paracetamol, saturan sus otras dos vías metabólicas y se crea un exceso de N-acetilbenzoquinoneimida que agota los niveles hepáticos de glutatión. Entonces el metabolito puede reaccionar covalentemente con aminoácidos de las enzimas y proteínas hepáticas, a las que inactiva y llega a provocar necrosis hepática aguda. Los niños tienen una menor capacidad de glucuronidación, lo que los hace más susceptibles a sufrir este trastorno. La eliminación es principalmente urinaria. El 90% de la dosis ingerida la elimina el riñón en 24 horas, principalmente como glucurónidos (60 a 80%) y sulfoconjugados (20 a 30%). Menos del 5% se elimina sin modificar. La semi-vida de eliminación del paracetamol es de 2-4 horas en los pacientes con la función hepática normal, siendo prácticamente indetectable en el plasma 8 horas después de su administración. En los pacientes con disfunción hepática la semi-vida aumenta sustancialmente, lo que puede ocasionar el desarrollo de una necrosis hepática.[5]
Toxicidad
Consideraciones generales
El paracetamol tiene un índice terapéutico muy ajustado. Esto significa que la dosis normal es cercana a la sobredosis, haciendo de él un compuesto peligroso. Una dosis única de paracetamol de 10 gramos o dosis continuadas de 5 g/día en un no consumidor de alcohol con buena salud, o 4 g/día en un consumidor habitual de alcohol, pueden causar daños importantes en el hígado. Sin un tratamiento adecuado en el momento oportuno, la sobredosis o la dosis casi normal de paracetamol puede dar como resultado un fallo hepático seguido de la muerte inevitable. Debido a la amplia disponibilidad, sin receta, del paracetamol, éste se ha utilizado en muchos intentos de suicidio.[6]
El paracetamol no debe tomarse tras consumir alcohol, debido a que el hígado, cuando está metabolizando el alcohol, no puede metabolizar simultáneamente el paracetamol, aumentando por tanto el riesgo de hepatotoxicidad. Usado responsablemente, el paracetamol es uno de los tratamientos más seguros disponibles para la analgesia. El compuesto carece de efectos sobre el sistema de la ciclooxigenasa, por lo tanto no tiene efectos negativos sobre el esófago, estómago, intestino delgado o intestino grueso, al contrario que los AINEs. Además, los pacientes con enfermedades del riñón pueden tomar paracetamol mientras que los AINEs pueden provocar insuficiencia renal aguda a ciertos pacientes. Además, el paracetamol tiene pocos problemas de interacción con otros medicamentos.
La potencia del analgésico es equivalente, cuando no hay inflamación, a la de los AINEs, siempre que la dosis de paracetamol sea la adecuada. Un gramo diario de paracetamol tiene un efecto analgésico equivalente al de los AINEs, por ejemplo en osteoartritis. Cuando se administra simultáneamente con 50 mg del antidepresivo tricíclico amitriptilina dos veces al día, dicha combinación es tan eficaz como la de paracetamol y codeína, pero no pierde efectividad con el tiempo como ocurre con la administración crónica de narcóticos. A diferencia del ácido acetilsalicílico, el paracetamol no contribuye al síndrome de Reye en niños con enfermedades víricas. Estos factores han hecho del paracetamol el analgésico preferido para pacientes hospitalizados en casos de dolor suave a moderado, además de ser el analgésico más utilizado en pacientes ambulatorios.
El paracetamol es extremadamente tóxico para los perros, gatos, etc. y no debería serles administrado bajo ninguna circunstancia.[7] Cualquier caso sospechoso debería ser tratado por un veterinario para proceder a la desintoxicación.[8] El tratamiento en gatos es muy similar al de los humanos. El acetaminophen es el calmante de dolor más ampliamente usado en los Estados Unidos (TYLENOL), pero puede destruir al hígado en dosis ordinarias o cercanas a ordinarias. Ese hecho será una noticia para muchos consumidores pero algo sabido para los especialistas en hígado, y ahora se reconoció por fin oficialmente en abril de 2009, por la Administración de Drogas y Alimentos (FDA, por sus siglas en inglés). El comité consultivo de la FDA declaró hace 32 años: "No se debe exceder la dosificación recomendada [acetaminophen-Tylenol] porque puede causar lesión hepática severa e incluso la muerte".[cita requerida] (Informes más completos se pueden encontrar en: worst pills y en la pág. de la FDA, U S Food and Drug Administration Home Page )
Mecanismo de la toxicidad
Como se mencionó anteriormente, el paracetamol es metabolizado a compuestos inactivos por combinación con sulfato y ácido glucurónico, siendo una pequeña parte metabolizada por el sistema del citocromo P-450. Éste oxida al paracetamol para producir un intermedio muy reactivo, la imina N-acetil-p-benzoquinoneimina (NAPQI). En condiciones normales, la NAPQI se neutraliza por acción del glutatión.
En episodios de toxicidad por paracetamol, las vías metabólicas del sulfato y la glucuronida se saturan y mayor cantidad paracetamol se desvía al sistema del citocromo P-450 donde se produce NAPQI. Consecuentemente, los suministros hepatocelulares de glutatión se agotan y en NAPQI puede reaccionar libremente con las membranas celulares, causando amplios daños y muerte de muchos hepatocitos, dando como resultado necrosis hepática aguda. En estudios en animales, debe consumirse el 70% del glutatión hepático antes de que se dé hepatotoxicidad.
Factores de riesgo
La dosis tóxica de paracetamol es muy variable. En adultos, dosis únicas por encima de 7,5 gramos o 150 mg/kg tienen una probabilidad razonable de causar hepatotoxicidad. En adultos, dosis de más de 25 gramos son potencialmente letales. También puede darse hepatotoxicidad cuando dosis pequeñas pero múltiples superan dichas cantidades en 24 horas, o mediante ingesta crónica de pequeñas dosis. Sin embargo, la sobredosis involuntaria de paracetamol en niños raramente tiene como resultado este tipo de toxicidad. Este hecho podría atribuirse en parte al sistema del citocromo P-450, inmaduro aún en los niños. El consumo excesivo de alcohol puede afectar la función renal e incrementar la toxicidad del paracetamol. Por esta razón, tras grandes ingestas de alcohol se recomiendan otros analgésicos como el ibuprofeno o el ácido acetilsalicílico.
Algunas personas son más propensas a la hepatotoxicidad, incluso con dosis bajas como 4 gramos/día, y las dosis letales pueden bajar hasta 6 g/día. El ayuno es un factor de riesgo, posiblemente debido a la reducción de las reservas de glutatión del hígado. Está bien documentado que el uso en combinación con de inductores del CYP2E1 como la isoniazida potencia la hepatotoxicidad, aunque dicha relación no está del todo clara.[9] [10] El alcoholismo, que también da como resultado la producción de CYP2E1, también aumenta el riesgo de hepatotoxicidad del paracetamol.[11] El uso en combinación con de otros medicamentos que inducen la síntesis de enzimas CYP, como los antiepilépticos (carbamazepina, fenitoína, barbitúricos, etc) también han sido presentados como factores de riesgo.
Evolución
Las personas que han ingerido una sobredosis de paracetamol, por lo general no presentan síntomas durante las primeras 24 horas. Aunque inicialmente son síntomas comunes náuseas, vómitos y diaforesis, éstos remiten pasadas varias horas. Tras aliviarse estos síntomas, los pacientes experimentan una mejoría, pudiendo llegar a pensar que lo peor ha pasado. Sin embargo, tras ingerir una dosis tóxica, tras estos síntomas se produciría un fallo hepático.
El daño se da generalmente en los hepatocitos a medida que van metabolizando el paracetamol. Sin embargo, también puede darse insuficiencia renal aguda. Esto también es causado generalmente por un fallo hepatorenal o por un fallo orgánico múltiple. La manifestación clínica principal de la intoxicación también podría ser la insuficiencia renal aguda. En estos casos, es posible que el metabolito tóxico sea producido en más cantidad en los riñones que en el hígado.
El pronóstico de la sobredosis por paracetamol varía dependiendo de la dosis y el tratamiento empleado. En algunos casos, la necrosis hepática masiva da como resultado un fallo hepático fulminante, con complicaciones como hemorragias, hipoglucemia, insuficiencia renal, encefalopatía hepática, edema cerebral, sepsis, fallo orgánico múltiple y muerte en pocos días. En muchos casos, la necrosis hepática puede continuar, volver la función hepática normal y el paciente puede sobrevivir con una función hepática normal en un plazo de pocas semanas.
Diagnóstico
Los síntomas claros de toxicidad hepática pueden sobrevenir en un plazo de 1 a 4 días, aunque en algunos casos éstos pueden ser evidentes en tan sólo 12 horas. Pueden darse molestias en el cuadrante derecho superior. Mediante análisis se puede determinar la existencia de necrosis hepática masiva si se detectan niveles elevados de AST, ALT, bilirrubina y tiempos de coagulación elevados (concretamente, tiempos elevados de protrombina). La hepatotoxicidad del paracetamol se puede diagnosticar si después de una sobredosis de paracetamol el AST y ALT superan los 1.000 UI/L. Sin embargo, los niveles de AST y ALT pueden superar los 10.000 UI/L. En general, en la hepatotoxicidad inducida por paracetamol, los niveles de AST son algo superiores a los de ALT.
Existen nomogramas de paracetamol que permiten estimar el riesgo de toxicidad basándose en su concentración en suero sanguíneo tras un determinado número de horas. Para determinar el riesgo potencial de hepatotoxicidad, el nivel de paracetamol debe ser seguido a lo largo del nomograma estándar. Un nivel de paracetamol trazado durante las primeras horas de ingestión podría subestimar la cantidad real en el organismo, debido a que en ese momento el paracetamol podría estar aún absorbiéndose en el tracto gastrointestinal. Una demora en la determinación del nivel de paracetamol en el organismo no es recomendable, debido a que en estos casos las estimaciones podrían no ser adecuadas y un nivel tóxico en cualquier momento es suficiente para administrar el antídoto.
Sobredosificación
Medidas de rescate
- Lavado gástrico
El tratamiento para sobredosis de paracetamol, sin complicaciones, es similar al usado en otros medicamentos, un lavado gastrointestinal. Adicionalmente, administrar N-acetilcisteína, ya sea por vía intravenosa u oral, ayuda mucho en estos casos. Hay suficiente margen para que el médico juzgue en este caso si es necesario un lavado gastrointestinal completo o basta con administrar carbón activado. La absorción total del paracetamol por parte del tracto gastrointestinal se completa en aproximadamente dos horas. En estos casos, el jarabe de ipecacuana (un emético) no es efectivo, debido a que induce vómitos y esto lo único que hace es retrasar la efectividad del carbón activado y la N-acetilcisteína, al tener que administrarlos después de que finalicen los vómitos. El lavado gástrico es efectivo durante la 1º hora posterior a la ingestión. Posterior a eso, no tiene utilidad clínica.
- Carbón activado
Normalmente, la administración de carbón activado es más efectiva que el lavado gástrico. Éste absorbe bien el paracetamol, y por lo tanto se reduce la cantidad que se absorbe en el tracto gastrointestinal. Además, también plantea menos riesgo de aspiración que el lavado gástrico. Hace tiempo había cierta renuencia a administrar carbón activado, debido al temor a que también absorbiese la N-acetilcisteína. Estudios recientes han demostrado que la cantidad absorbida por esta vía no supera el 39% cuando ambos se administran conjuntamente. Otros estudios han mostrado que el carbón activado parece ser beneficioso para el paciente. Hay un consenso general en administrar carbón activado durante las primeras 4 horas tras la sobredosis; tras este tiempo, depende del criterio del médico, pero de todas formas se considera un tratamiento benigno. Si hay dudas sobre la ingestión de paracetamol junto a otros medicamentos, entonces debe administrarse carbón activado. Hay discrepancias en cuanto a cambiar la dosis de N-acetilcisteína administrada, o incluso si ésta debe modificarse.
La dosis de carbón activado es de 2 gr/kg de peso del paciente hasta un tope de 100 gramos totales. En niños, la dosis es de 1 gr/kg. Se administra por vía oral y puede ser junto a agua o jugo para enmascarar en parte el mal sabor de éste.
Acetilcisteína
La N-acetilcisteína (NAC) actúa proporcionando grupos sulfhidrilo para que reaccionen con el metabolito tóxico y de esta forma no ataquen a los hepatocitos. Si la NAC se administra en las primeras ocho horas, se reduce notablemente la toxicidad. Si se administra pasadas 8 horas, su eficacia se reduce debido a que ha ya empezado la cascada de reacciones tóxicas en el hígado, y el riesgo de necrosis hepática aumenta considerablemente. La NAC oral es un medicamento seguro, es fiable en casos de sobredosis por paracetamol durante el embarazo y no se dan reacciones adversas con pronóstico fatal. El fabricante recomienda no administrar el NAC si existe una encefalopatía, debido a que existen razones teóricas que arguyen que dicha encefalopatía podría empeorar. A principios de 2004, la Administración de Drogas y Alimentos (FDA, por sus siglas en inglés) estadounidense autorizó el uso, para pacientes con sobredosis de más de 10 horas, de un preparado de NAC para infusión intravenosa (dosis total de 300 mg/kg) durante un período de 20 horas, que carece de efectos pirogénicos. Este preparado se ha usado con éxito durante años en otros países, como Australia, Canadá y Gran Bretaña.
- Administración
Dicho tratamiento consiste en una administración inicial de 150 mg/kg durante 15 minutos, seguido de 50 mg/kg durante las cuatro horas siguientes y finalmente 100 mg/kg durante las 16 horas restantes. La formulación oral también puede disolverse, filtrarse y esterilizarse por un farmacéutico del hospital para administración intravenosa. Ésta es una buena opción en casos donde la vía enteral no es viable o está contraindicada. La administración intravenosa de NAC está relacionada con casos de reacciones alérgicas como choques anafilácticos y broncoespasmos.
En la práctica, si han transcurrido más de 8 horas tras la ingestión, el carbón activado no es efectivo y debe administrarse la NAC inmediatamente. Si han transcurrido menos de 8 horas, se debe administrar carbón activado, empezar a administrar NAC y esperar a ver los niveles de paracetamol. En pacientes con una sobredosis de menos de 8 horas, el riesgo de hepatotoxicidad es reducido. Si se administran más de dos dosis de carbón activado debido a que el paciente ha ingerido dos o más medicamentos, las subsiguientes administraciones de carbón activado y NAC deben demorarse dos horas. La NAC es eficaz si se administra con prontitud, pero puede ser efectivo aún cuando hayan transcurrido 48 horas de la sobredosis.
En general, la NAC oral se administra enteralmente con una primera dosis de 140 mg/kg seguidas de 17 dosis más, cada cuatro horas, de 40 mg/kg. La NAC puede ser difícil de administrar debido a su sabor y es frecuente que provoque vómitos y náuseas. Para maximizar su tolerancia, puede diluirse del 5% al 20% a partir de las dosis comerciales. Los estudios iniciales de laboratorio deben incluir bilirrubina, AST, ALT y el tiempo de protrombina. Los análisis deben repetirse, al menos, diariamente. Una vez que se ha determinado que se ha ingerido una dosis potencialmente tóxica, deben administrarse las 17 dosis de NAC, aunque el paracetamol devenga indetectable en sangre. Si se desarrolla un fallo hepático, deben continuarse las 17 dosis hasta que se restablezca la función hepática normal o se efectúe un trasplante de hígado.
Pronóstico
El riesgo de mortalidad por sobredosis empieza a aumentar a partir de los dos días, alcanza un máximo a los cuatro y posteriormente disminuye gradualmente. Los pacientes con una mala evolución deben ser trasladados inmediatamente a un centro capaz de efectuar trasplantes de hígado. La acidosis es el factor más ominoso que delata el riesgo de mortalidad y la necesidad de un trasplante.
En pacientes no trasplantados, se ha establecido un factor de mortalidad del 95% cuando el pH sanguíneo se sitúa por debajo de 7,3. Otros indicadores de un mal pronóstico médico incluyen insuficiencia renal, grado 3 o mayor de encefalopatía hepática, un tiempo de protrombina marcadamente elevado o un aumento en el mismo del día 3 al 4. Un estudio ha mostrado que un análisis del factor V menor que el 10% del normal indica un mal pronóstico (91% de mortalidad), mientras que una relación menor a 30 entre el factor VIII y el V son indicadores de un buen pronóstico (100% de supervivencia).
Incidencia ambiental y degradación
Uno de los fármacos más ampliamente encontrado y en mayores concentraciones es el paracetamol, producto que puede encontrarse en efluentes hospitalarios, efluentes de plantas de tratamiento, ríos y lodos. Lamentablemente los medicamentos son diseñados para que posean determinadas características, por ejemplo, aproximadamente el 30 % de los medicamentos son lipofílicos, que significa que se disuelven en grasa pero no en agua. Esta característica le permite a estos compuestos pasar a través de las membranas de la célula y actuar dentro de las mismas. Por otro lado, los medicamentos se diseñan para que sean persistentes, por lo que mantienen su estructura química un tiempo suficientemente grande como para ejercer su acción terapéutica, así que una vez que entran al medio ambiente persisten en el mismo.
El paracetamol en solución acuosa es susceptible de sufrir una hidrólisis para formar el p-amino fenol, el mismo es susceptible de degradarse en quinoneimina. La velocidad de degradación del paracetamol crece con el aumento de la temperatura y de la luz. Esta velocidad es mínima a un pH cercano a 6. Para estabilizar el paracetamol en solución, se suele añadir un tampón y un agente antioxidante o captador de radicales. En la eliminación del acetaminofeno se ha observado que reacciona con cloro para formar un gran número de subproductos, dos de los cuales han sido identificados como tóxicos. [12]
Véase también
- Analgésico
- Antipirético
- Cefalea y cefalea tensional
- Industria farmacéutica
- Medicina basada en la evidencia
- Toxicología
- Vademécum
Referencias
- ↑ Brodie BB & Axelrod J (1948). J. Pharmacol. Exp. Ther. 94: 29-38.
- ↑ Briefing Materials For Drug Safety And Risk Management Anesthetic And Life Support And Nonprescription Drugs Advisory Commitee Meeting
- ↑ Boutaud O, Aronoff DM, Richardson JH, Marnett LJ, Oates JA (2002). "Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H2 synthases". Proc Natl Acad Sci U S A 99 (10): 7130-5. PMID Disponible en línea.
- ↑ Swierkosz TA, Jordan L, McBride M, McGough K, Devlin J, Botting RM (2002). "Actions of paracetamol on cyclooxygenases in tissue and cell homogenates of mouse and rabbit". Med Sci Monit 8 (12): BR496-503. PMID PDF.
- ↑ Benson GD. Acetaminophen in chronic liver disease. Clin Pharmacol Ther 1983;33:95—101
- ↑ De la Oliva, S.; Cánovas, A.; Mencías, E. (2001). "Revisión de las intoxicaciones por paracetamol consultadas al Servicio de Información Toxicológica. Comunicación presentada en el XIV Congreso Español de Toxicología. Murcia, 26-28 de septiembre de 2001". Revista de Toxicología. ISSN: 1697-0748, 18 (3): 183. Disponible en línea.
- ↑ Allen AL (2003). "The diagnosis of acetaminophen toxicosis in a cat". Can Vet J 44 (6): 509-10. PMID Disponible en línea
- ↑ Villar D, Buck WB, Gonzalez JM (1998). "Ibuprofen, aspirin and acetaminophen toxicosis and treatment in dogs and cats". Vet Hum Toxicol 40 (3): 156-62. PMID
- ↑ Crippin JS (1993). "Acetaminophen hepatotoxicity: potentiation by isoniazid". Am J Gastroenterol 88 (4): 590-2. PMID
- ↑ Nolan CM, Sandblom RE, Thummel KE, Slattery JT, Nelson SD (1994). "Hepatotoxicity associated with acetaminophen usage in patients receiving multiple drug therapy for tuberculosis". Chest 105 (2): 408-11. PMID
- ↑ Zimmerman HJ, Maddrey WC (1995). "Acetaminophen (paracetamol) hepatotoxicity with regular intake of alcohol: analysis of instances of therapeutic misadventure". Hepatology 22 (3): 767-73. PMID
- ↑ «Formulación líquida de paracetamol inyectable - Una solución acuosa de paracetamol para uso por perfusión con un pH entre 4,5 y 6,0, caracterizada». Patentados.com (27-10-2007). Consultado el 19-03-2011.
Enlaces externos
 Wikimedia Commons alberga contenido multimedia sobre ParacetamolCommons.
Wikimedia Commons alberga contenido multimedia sobre ParacetamolCommons.- En MedlinePlus puede encontrar más información sobre Paracetamol
- En Medline hay más información sobre Paracetamol (en inglés)
- Paracetamol en Nomenclator
- Información detallada sobre Paracetamol en Vademecum.es
- U S Food and Drug Administration Home Page (en inglés)
- Tylenol (Sitio web oficial)
- "El Asesino del Tylenol" en "Escrito con Sangre", página sobre asesinos seriales
- En AEMPs
- Hepatotoxicidad por paracetamol
- Paracetamol Information Centre (en inglés)
- History of paracetamol (en inglés)
- US Patent 6,126,967 (en inglés)
- History of paracetamol (en inglés)
- History & chemistry of paracetamol (en inglés)
- The Julius Axelrod Papers (en inglés)
- Paracetamol Side Effects (en inglés)
- Paracetamol
- Mecanismo de acción del paracetamol
Categorías:- Código ATC N
- Amidas
- Analgésicos-antipiréticos no opioides




Wikimedia foundation. 2010.